Conservation vs Fitness -- Linking FB Genes to Pangenome Clusters
CompletedResearch Question
Are essential genes preferentially conserved in the core genome, and what functional categories distinguish essential-core from essential-auxiliary genes?
Research Plan
Hypothesis
The Fitness Browser provides mutant fitness data for ~221K genes across 48 bacteria, while the KBase pangenome classifies 132.5M gene clusters by conservation level (how many genomes in a species carry a given cluster). By linking these datasets, we can ask: do core genes (present in >=95% of species genomes) show different fitness patterns than auxiliary or singleton genes? We expect essential genes to be enriched in the core genome, and that functional profiles will differ between essential-core and essential-auxiliary genes.
Approach
- Link table (Phase 1) -- Map FB genes to pangenome clusters via DIAMOND blastp (>=90% identity, best hit per gene), resolving GTDB taxonomic renames via three matching strategies (NCBI taxid, organism name, scaffold accession)
- Essential gene identification (Phase 2) -- Identify putative essential genes as protein-coding genes (type=1) absent from
genefitness(no viable transposon mutants recovered) - Conservation analysis -- Compare conservation status (core/auxiliary/singleton) between essential and non-essential genes across 34 organisms
- Functional characterization -- Use FB annotations (SEED, KEGG) to profile essential genes by conservation category
Revision History
- v1 (2026-02): Migrated from README.md
Overview
This project builds the bridge between the Fitness Browser (~221K genes, 48 bacteria) and the KBase pangenome (132.5M gene clusters). It maps FB genes to pangenome clusters via DIAMOND blastp, identifies putative essential genes (no viable transposon mutants), and tests whether essential genes are enriched in core clusters. It also profiles essential genes by conservation category, revealing that essential-core genes are enzyme-rich and well-annotated while essential-auxiliary genes are poorly characterized.
Key Findings
Link Table (Phase 1)
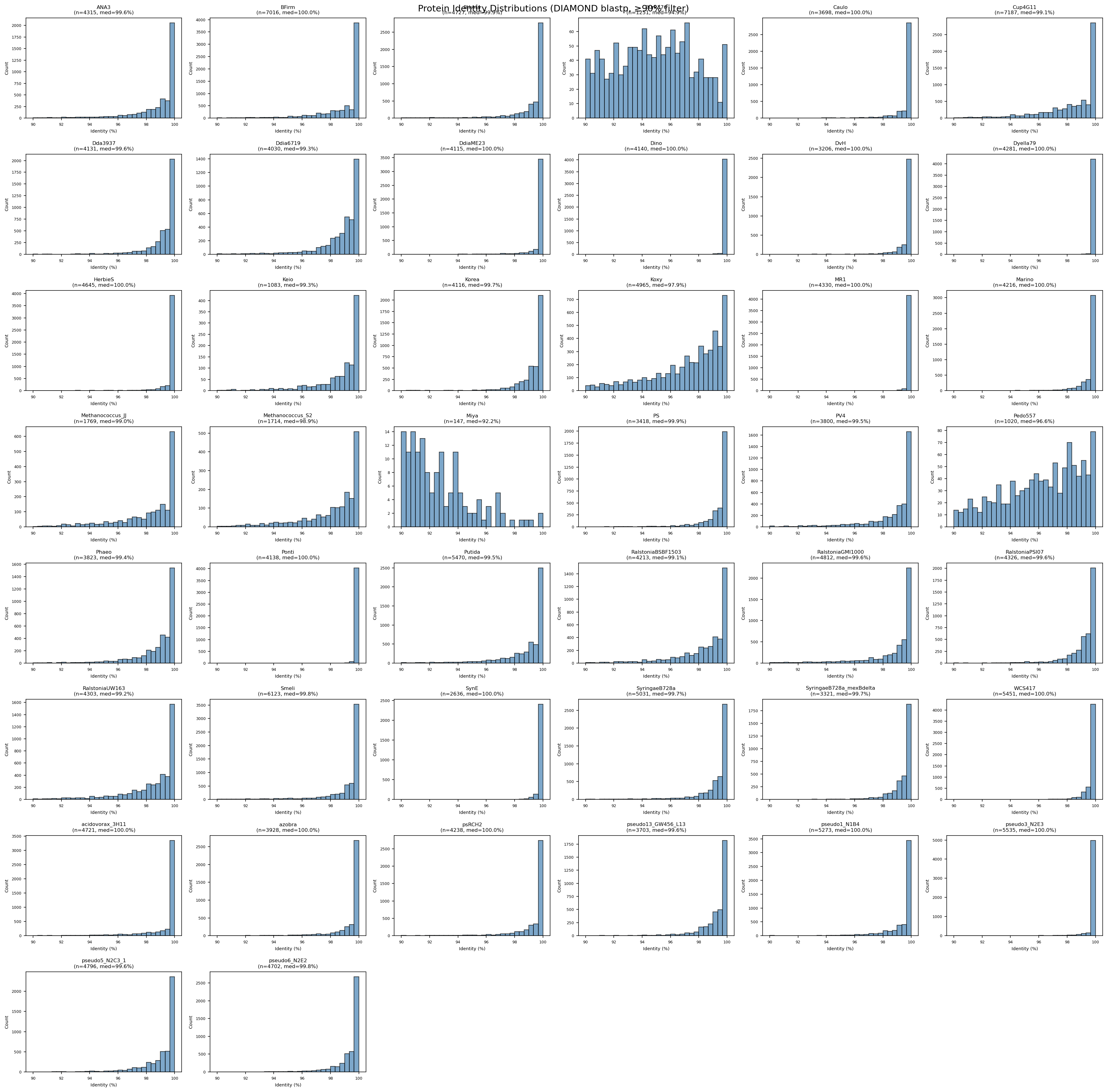
- 44 of 48 FB organisms mapped to pangenome species clades
- 177,863 gene-to-cluster links at 100.0% median protein identity, 94.2% median gene coverage
- 34 organisms have >=90% coverage; 33 used for downstream analysis (Dyella79 excluded due to locus tag mismatch)
- 4 organisms unmatched: Cola, Kang, Magneto, SB2B (species had too few genomes in GTDB for pangenome construction)
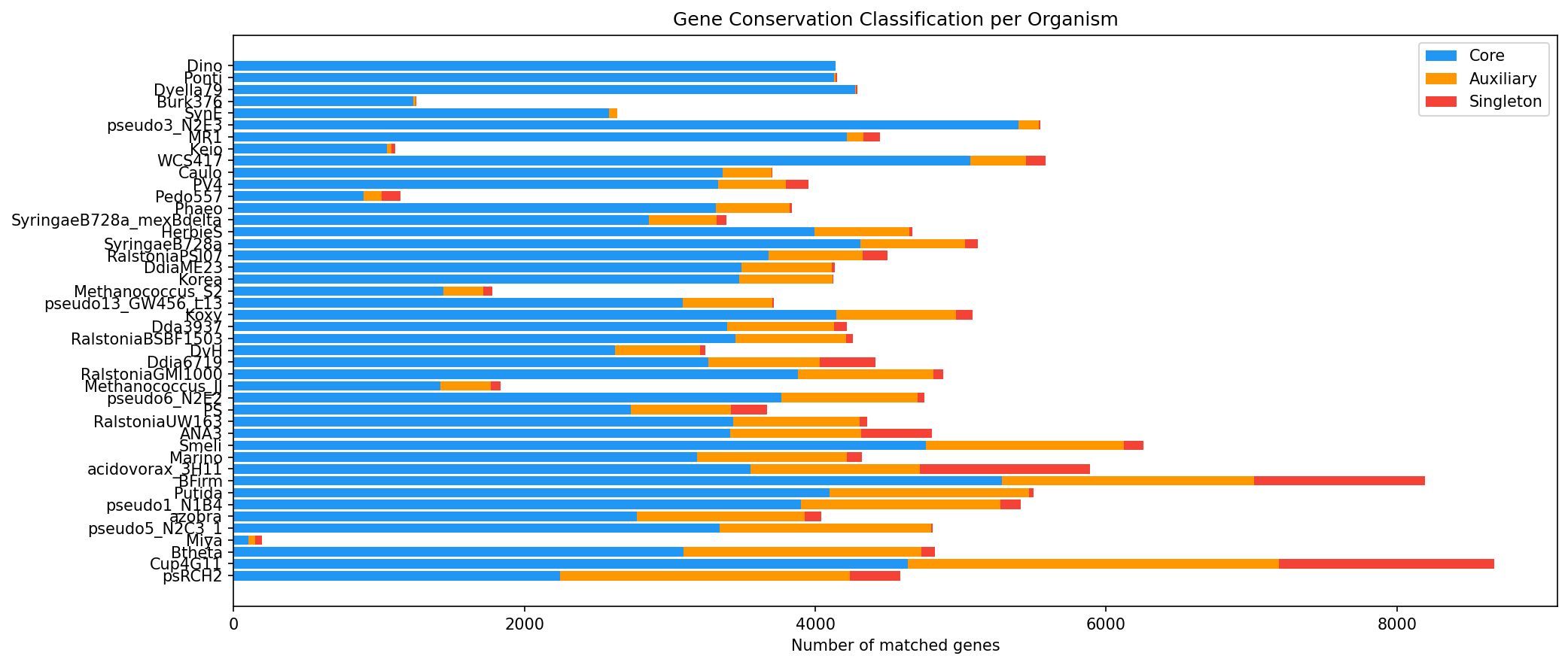
- Conservation breakdown: 145,821 core (82.0%), 32,042 auxiliary (18.0%) -- of which 7,574 are singletons (singletons are a subset of auxiliary)
(Notebook: 01_organism_mapping.ipynb, 03_build_link_table.ipynb)
Essential Genes Are Enriched in Core Clusters (Phase 2)
- 27,693 putative essential genes identified (18.6% of 148,826 protein-coding genes across 33 organisms; range 12.9-28.9% per organism)
- Essential genes are 86.1% core vs 81.2% for non-essential genes
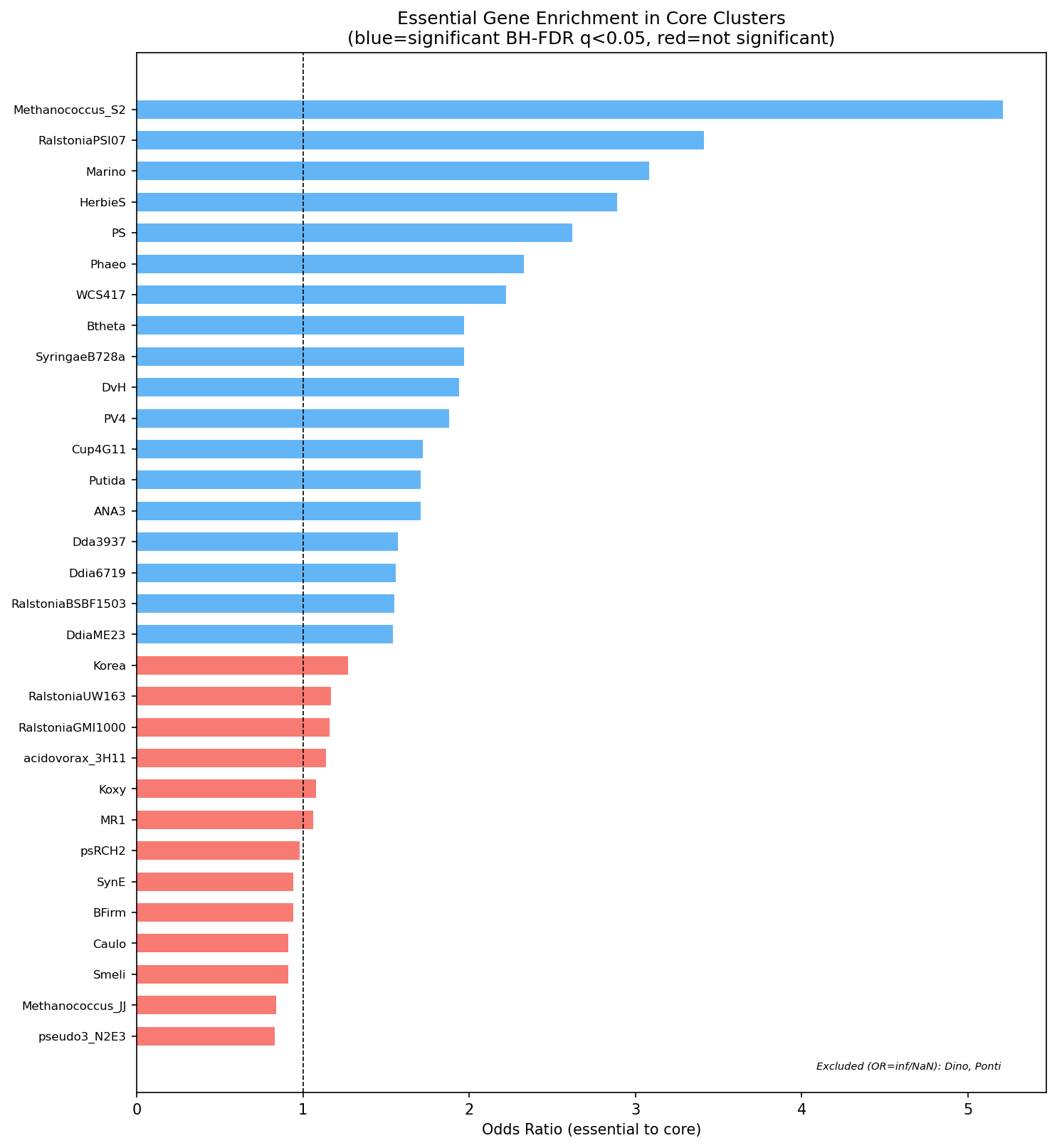
- Median odds ratio 1.56 -- essential genes are 1.56x more likely to be in the core genome
- 18 of 33 organisms show statistically significant enrichment (Fisher's exact test, BH-FDR q < 0.05)
- Strongest signal: Methanococcus maripaludis S2 (OR=5.21), Ralstonia syzygii PSI07 (OR=3.41), Marinobacter adhaerens (OR=3.08)
(Notebook: 04_essential_conservation.ipynb)
Functional Profiles Differ by Conservation Category
| Category | n genes | % Enzyme | % Hypothetical |
|---|---|---|---|
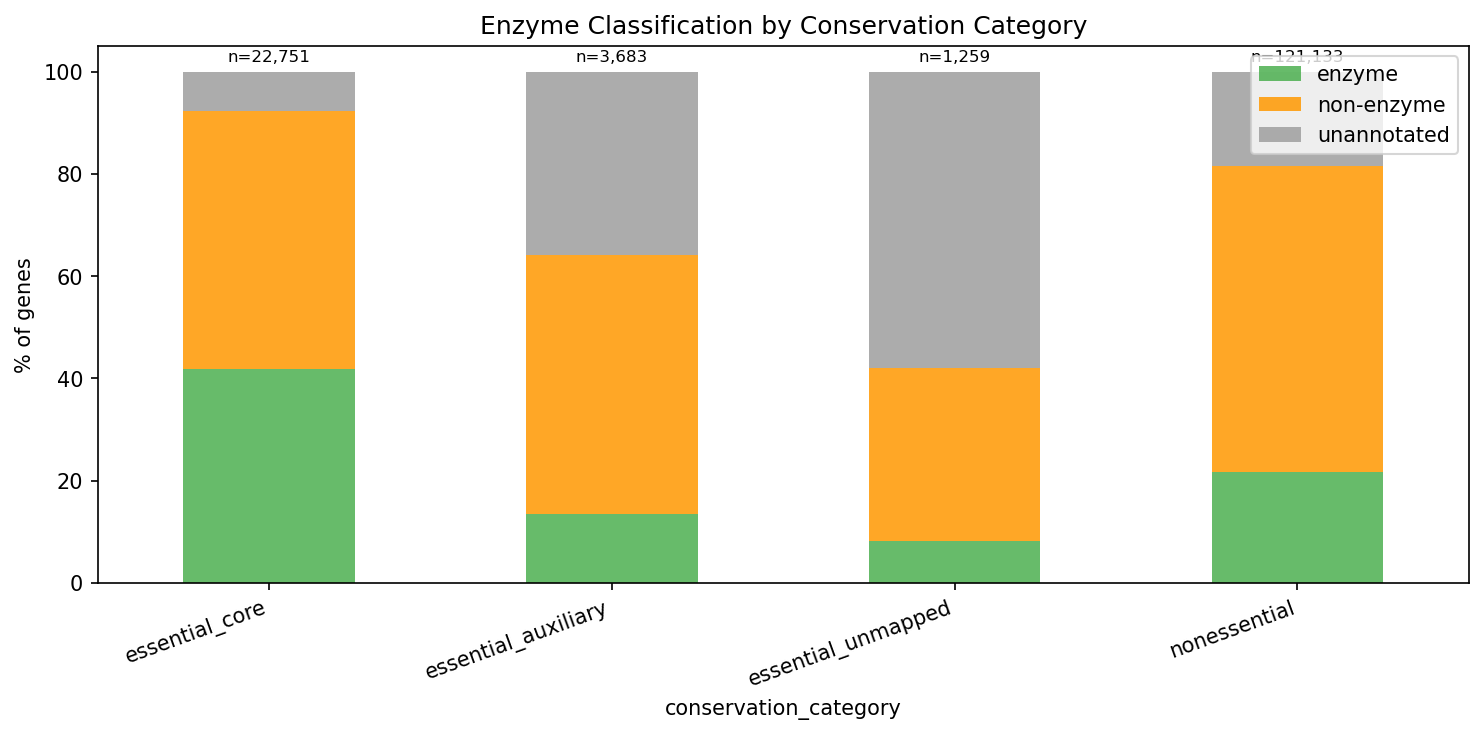
| Essential-core | 22,751 | 41.9% | 13.0% |
| Essential-auxiliary | 3,683 | 13.4% | 38.2% |
| Essential-unmapped | 1,259 | 18.2% | 44.7% |
| Non-essential | 124,744 | 21.5% | 24.5% |
Essential-core genes are the most enzyme-rich (41.9%) and best-annotated (87% with known function). They are enriched in Protein Metabolism (+13.7 percentage points vs non-essential), Cofactors/Vitamins (+6.2%), Cell Wall (+3.9%), and Fatty Acid biosynthesis (+3.1%). They are depleted in Carbohydrates (-7.9%), Amino Acids (-5.6%), and Membrane Transport (-4.0%) -- functions that tend to be conditionally important rather than universally essential.
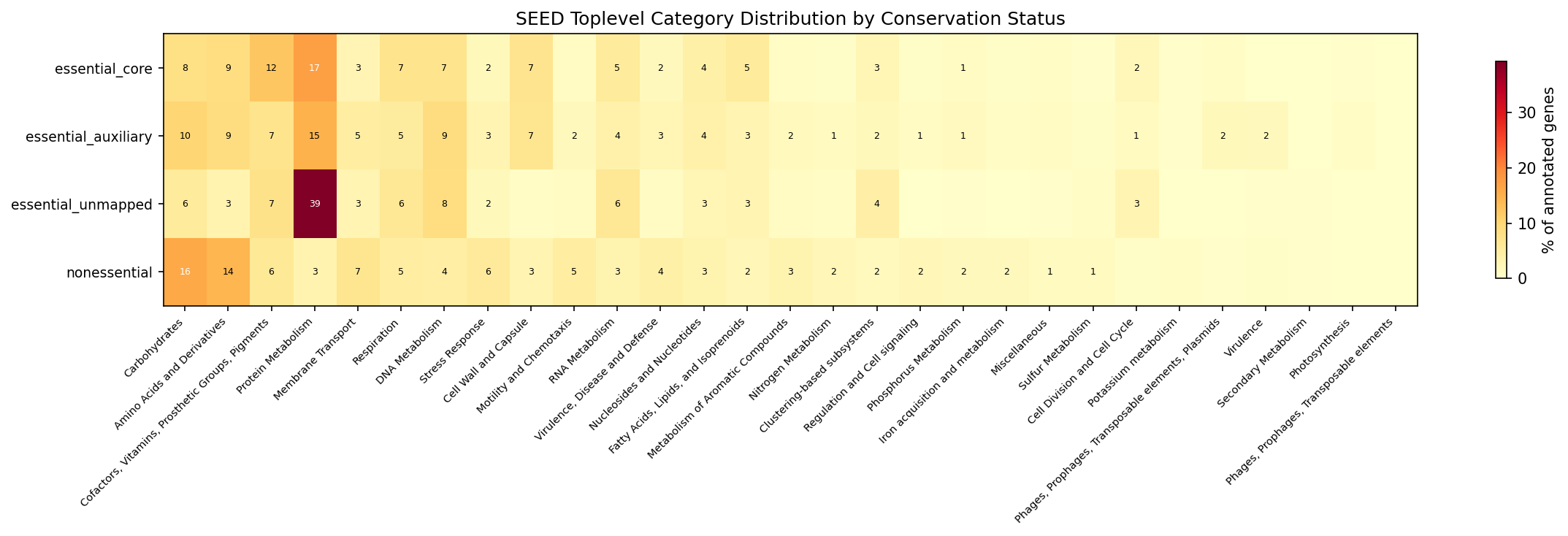
(Notebook: 04_essential_conservation.ipynb)
Essential-auxiliary genes (3,683 genes essential for viability but not in all strains) are poorly characterized (38.2% hypothetical) and less likely to be enzymes (13.4%). Top subsystems: ribosomes, DNA replication, type 4 secretion, plasmid replication -- suggesting strain-specific variants of core machinery plus mobile genetic elements.
Essential-unmapped genes (1,259 strain-specific essentials with no pangenome cluster match) are the least characterized (44.7% hypothetical). Known functions include divergent ribosomal proteins (L34, L36, S11, S12), translation factors, transposases, and DNA-binding proteins -- likely recently acquired or highly divergent variants of core functions.
Validation
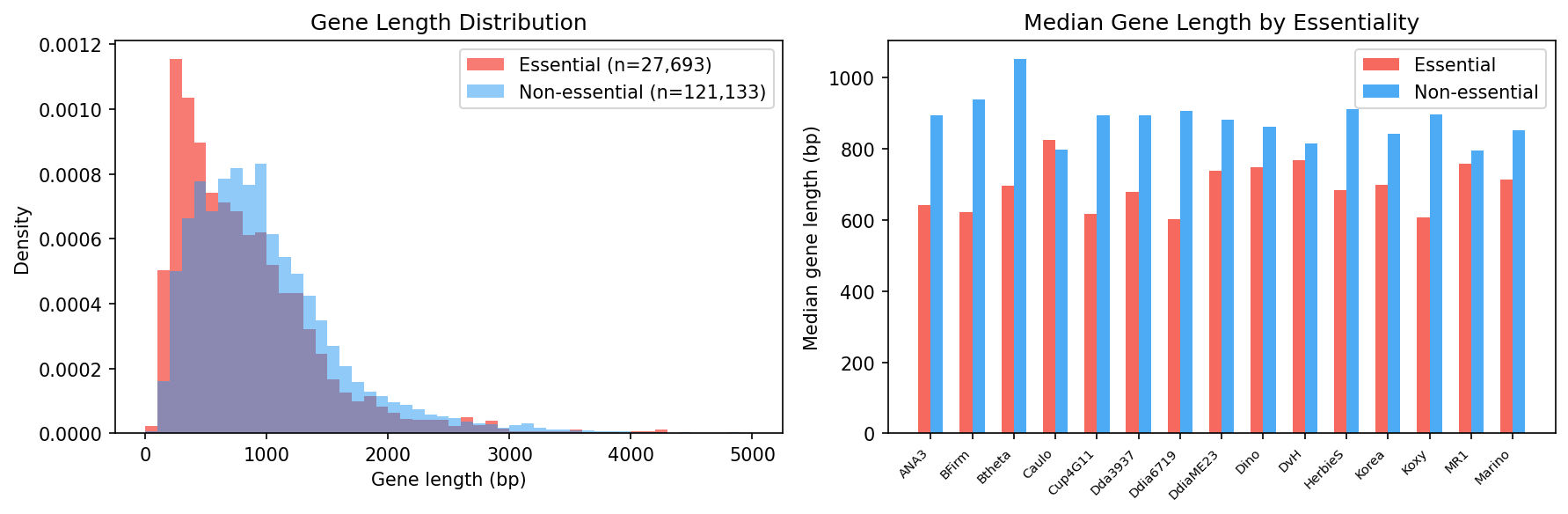
Gene length validation confirms that essential genes are slightly shorter on average, consistent with some insertion bias in transposon data.
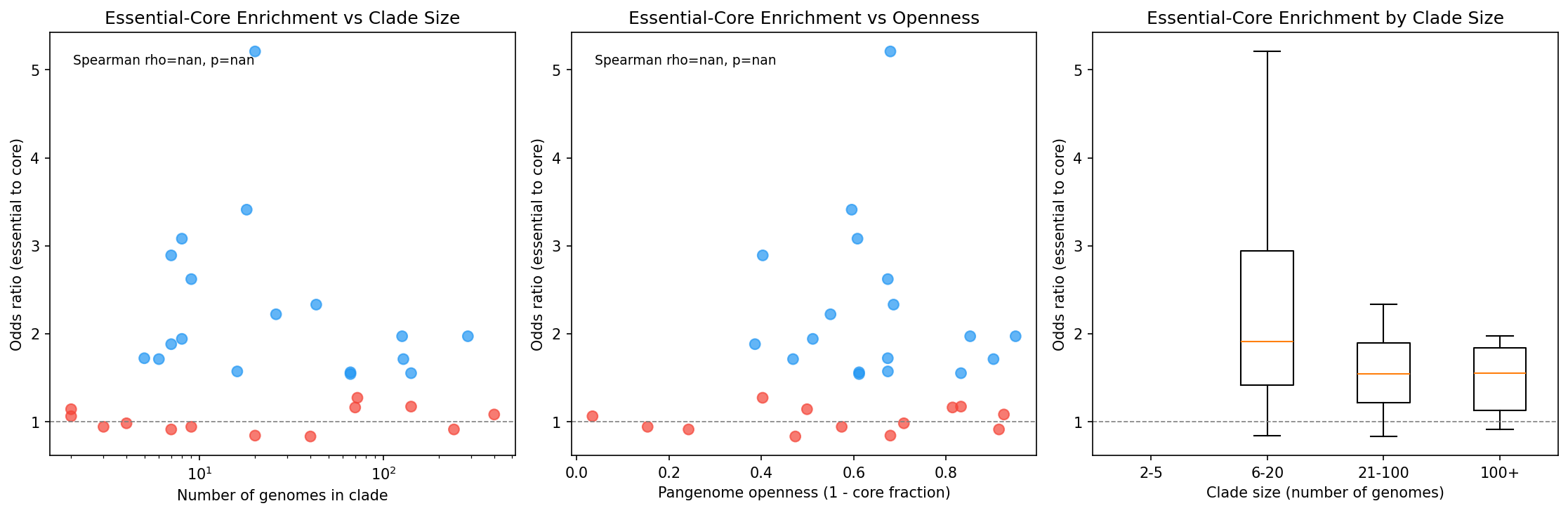
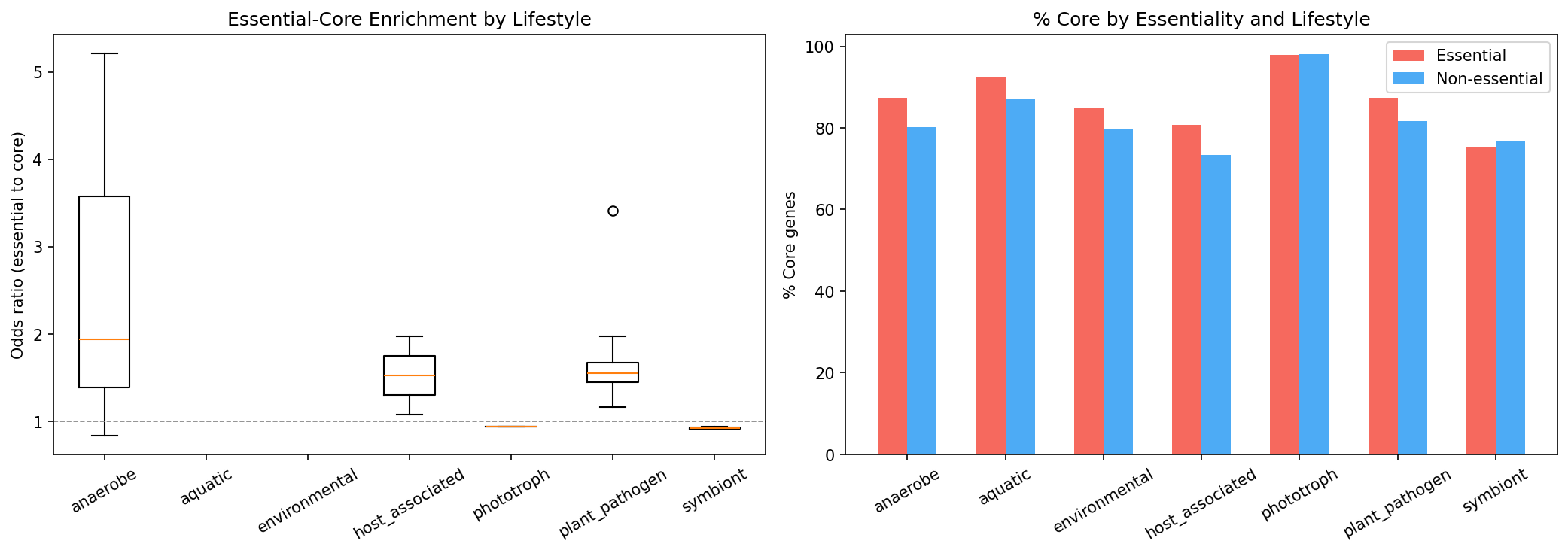
Clade size and lifestyle stratification show that the essential-core enrichment is robust across diverse genomic contexts.
(Notebook: 04_essential_conservation.ipynb)
Interpretation
Literature Context
- Our finding that essential genes are enriched in core clusters aligns with the general expectation that genes required for viability are conserved across a species. However, the enrichment is modest (OR=1.56), reflecting that most genes in well-characterized bacteria are core regardless of essentiality.
- Rosconi et al. (2022) studied essentiality across 36 S. pneumoniae strains and found that the pan-genome makes gene essentiality strain-dependent. They identified "universal essential," "core strain-specific essential," and "accessory essential" genes -- directly paralleling our essential-core, essential-auxiliary, and essential-unmapped categories. Our work extends this concept across 33 diverse bacterial species.
- Hutchison et al. (2016) designed a minimal Mycoplasma genome (473 genes) and found that 149 essential genes (31%) had unknown function. This parallels our finding that essential-unmapped genes are 44.7% hypothetical -- essential genes remain among the least characterized.
- Goodall et al. (2018) used TraDIS to define the E. coli K-12 essential genome and noted that gene length biases can affect essentiality calls from transposon data. Our gene length validation (NB04) addresses this concern.
- Price et al. (2018) generated the Fitness Browser data used here, demonstrating genome-wide mutant fitness across 32 bacteria. Our work adds a pangenome conservation dimension to their fitness data.
Limitations
- Essential gene definition is an upper bound: Genes without fitness data may lack transposon insertions due to being short, in low-complexity regions, or at scaffold edges -- not necessarily because they are essential. Gene length validation shows essential genes are slightly shorter on average, suggesting some insertion bias.
- Pangenome coverage varies: Clades with only 2 genomes have trivially high core fractions (a gene in both genomes = 100% = core), reducing the discriminative power of core/auxiliary classification.
- E. coli excluded: The main E. coli clade was absent from the pangenome (too many genomes). Keio (E. coli BW25113) mapped to the small
s__Escherichia_coli_Eclade at only 26.1% coverage. - Single growth condition for essentiality: RB-TnSeq essentiality is defined under the specific library construction conditions. Genes essential only under stress conditions would not be captured.
- Dyella79 excluded from Phase 2 due to locus tag format mismatch between FB gene table (
N515DRAFT_*) and protein sequences (ABZR86_RS*), causing 0% join rate. - 10 organisms excluded from Phase 2 due to <90% DIAMOND coverage, reducing taxonomic breadth.
Future Directions
- Condition-specific fitness vs conservation: Extend beyond essentiality to ask whether genes important for specific stress conditions (fitness < -2) show different conservation patterns
- Cross-organism essential gene families: Use FB ortholog data to identify essential gene families conserved across multiple species
- Quantitative fitness vs conservation: Correlate mean fitness effect (not just essential/non-essential binary) with core genome fraction
- Accessory genome essential functions: Deeper characterization of the 3,683 essential-auxiliary genes -- are they compensating for missing core functions?
Data
Sources
| Dataset | Description | Source |
|---|---|---|
| Fitness Browser gene table | ~221K genes across 48 bacteria | Price et al. (2018) |
| KBase pangenome clusters | 132.5M gene clusters across 27,690 species | Parks et al. (2022) |
| DIAMOND blastp | Protein similarity search for gene-to-cluster mapping | Buchfink et al. (2015) |
| SEED annotations | Functional category assignments | Overbeek et al. (2014) |
Generated Data
| File | Description |
|---|---|
data/organism_mapping.tsv |
FB org to clade mapping (44 organisms) |
data/fb_pangenome_link.tsv |
Final link table (177,863 rows) |
data/essential_genes.tsv |
Gene essentiality classification (153,143 genes) |
References
- Price MN et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557:503-509. DOI: 10.1038/s41586-018-0124-0. PMID: 29769716
- Parks DH et al. (2022). "GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy." Nucleic Acids Res 50:D199-D207. PMID: 34520557
- Rosconi F et al. (2022). "A bacterial pan-genome makes gene essentiality strain-dependent and evolvable." Nat Microbiol 7:1580-1592. DOI: 10.1038/s41564-022-01208-7. PMID: 36097170
- Hutchison CA 3rd et al. (2016). "Design and synthesis of a minimal bacterial genome." Science 351:aad6253. DOI: 10.1126/science.aad6253. PMID: 27013737
- Goodall ECA et al. (2018). "The Essential Genome of Escherichia coli K-12." mBio 9:e02096-17. DOI: 10.1128/mBio.02096-17. PMID: 29463657
- Deutschbauer A et al. (2014). "Towards an informative mutant phenotype for every bacterial gene." J Bacteriol 196:3643-55. DOI: 10.1128/JB.01836-14. PMID: 25112473
Discoveries
Across 33 diverse bacteria, putative essential genes (no transposon insertions in RB-TnSeq) are 86.1% core vs 81.2% for non-essential genes (median OR=1.56, 18/33 significant after BH-FDR). The enrichment is real but modest — most genes in well-characterized bacteria are core regardless of essential
Read more →Essential genes that map to core clusters are 41.9% enzymes and only 13.0% hypothetical — they are the well-characterized metabolic backbone (ribosomes, DNA replication, cell wall, cofactor biosynthesis). Essential-auxiliary genes (essential but not in all strains) are only 13.4% enzymes and 38.2% h
Read more →Essential-core genes are enriched in Protein Metabolism (+13.7 pp vs non-essential), Cofactors/Vitamins (+6.2 pp), Cell Wall (+3.9 pp). They are depleted in Carbohydrates (-7.9 pp), Amino Acids (-5.6 pp), Membrane Transport (-4.0 pp) — functions that are conditionally important rather than universal
Read more →The Fitness Browser aaseqs file (fit.genomics.lbl.gov/cgi_data/aaseqs) uses RefSeq-style locus tags (e.g., ABZR86_RS) for some organisms, while the FB gene table uses the original annotation locus tags (e.g., N515DRAFT_). This caused a complete join failure for Dyella79 (0% merge rate). Only 1 o
Data Collections
Used By
Data from this project is used by other projects.
Pan-Bacterial Metal Fitness Atlas
View project →
Core Gene Paradox -- Why Are Core Genes More Burdensome?
View project →
Lab Fitness Predicts Field Ecology at Oak Ridge
View project →
Pan-Bacterial AMR Gene Landscape
View project →
Metabolic Capability vs Dependency
View project →
Metabolic Capability vs Metabolic Dependency
View project →
Fitness Effects vs Conservation -- Quantitative Analysis
View project →
Gene Conservation, Fitness, and the Architecture of Bacterial Genomes
View project →
Co-fitness Predicts Co-inheritance in Bacterial Pangenomes
View project →
Field vs Lab Gene Importance in Desulfovibrio vulgaris Hildenborough
View project →
The Pan-Bacterial Essential Genome
View project →
The 5,526 Costly + Dispensable Genes
View project →
Fitness Modules x Pangenome Conservation
View project →
Truly Dark Genes — What Remains Unknown After Modern Annotation?
View project →
Review
Summary
This is an exceptionally well-executed cross-database analysis that bridges Fitness Browser RB-TnSeq essentiality data with KBase pangenome conservation classifications across 33 diverse bacteria. The project asks a clear, testable question — are essential genes preferentially conserved in the core genome? — and answers it with appropriate statistical rigor (Fisher's exact test with BH-FDR correction, median OR=1.56, 18/33 significant). The functional characterization adds genuine biological insight: essential-core genes are enzyme-rich and well-annotated (41.9% enzymes, 13.0% hypothetical), while essential-auxiliary and essential-unmapped genes represent poorly characterized frontiers (38–58% hypothetical). The project excels in pipeline architecture (clean Spark/local separation via src/ scripts with cached TSV intermediates), documentation (thorough README with quantitative results, literature context, candid limitations, and step-by-step reproduction guide), and pitfall awareness (correctly handles GTDB renames, FB string-typed columns, KEGG join paths, locus tag mismatches). The main areas for improvement are minor: NB01/NB02 lack saved outputs, a few README numbers don't match the NB04 actuals after Dyella79 exclusion, and the SEED hierarchy extraction step is documented but not integrated into the pipeline script.
Methodology
Research question: Clearly stated and testable. The two-phase structure (Phase 1: build the link table; Phase 2: essential genes vs conservation) is well-motivated and logically sequenced. The extension into functional characterization (Sections 6–8 of NB04) goes beyond the original question to provide genuinely useful biological insight.
Organism matching: The three-strategy approach (NCBI taxid → NCBI organism name → scaffold accession) is thorough and well-justified given GTDB taxonomic renames. The src/run_pipeline.py script properly escapes single quotes in organism names (lines 168–169), demonstrating attention to SQL safety. Multi-clade resolution by DIAMOND hit count is a reasonable heuristic, and the NB03 output confirms it works correctly — for example, Pseudomonas organisms mapped to multiple GTDB sub-clades are resolved to the best-fitting one.
Essential gene definition: Well-documented as an upper bound on true essentiality (type=1 CDS absent from genefitness). The NB04 header includes a thorough explanation of why this definition is necessary — after checking all 45 tables in kescience_fitnessbrowser, there is no explicit essentiality flag. The gene length validation (NB04 Section 1, Mann-Whitney U test) directly addresses the insertion bias concern raised by Goodall et al. (2018), and this caveat is honestly acknowledged in the README Limitations.
Statistical methods: Fisher's exact test per organism with Benjamini-Hochberg FDR correction is appropriate for the 2×2 contingency tables (essential/non-essential × core/non-core). Spearman correlations for pangenome context variables (clade size, openness) are suitable for non-normal distributions. The lifestyle stratification (Section 5) provides an additional biological dimension.
Data sources: Comprehensively documented in a README table covering 10 database tables plus external protein sequences. The KEGG annotation join path (besthitkegg → keggmember → kgroupdesc) correctly follows the pattern documented in docs/pitfalls.md, avoiding the known join-key gotcha.
Code Quality
SQL queries: Correct and efficient throughout. Key observations:
- The extract_essential_genes.py queries properly use orgId filters on the 27M-row genefitness table and cast coordinate columns with CAST(begin AS INT) / CAST(end AS INT), consistent with the documented pitfall that all FB columns are strings.
- Per-clade FASTA extraction in run_pipeline.py uses exact equality on the partitioned gtdb_species_clade_id column rather than LIKE patterns — consistent with performance guidance in docs/pitfalls.md.
- The KEGG join in extract_essential_genes.py (lines 120–128) correctly follows the three-table besthitkegg → keggmember → kgroupdesc path.
- The type = '1' filter (string comparison) for protein-coding genes is correct given the all-strings schema.
Pitfalls from docs/pitfalls.md addressed:
- ✅ String-typed numeric columns: Cast before comparison throughout
- ✅ FB gene table has no essentiality flag: Derives essentiality from absence in genefitness
- ✅ FB aaseqs locus tag mismatch: Dyella79 explicitly excluded with clear documentation
- ✅ KEGG annotation join path: Uses correct besthitkegg → keggmember → kgroupdesc chain
- ✅ Spark LIMIT/OFFSET pagination: Uses per-clade queries instead of pagination
- ✅ Core/auxiliary/singleton mutual exclusivity and subset relationship: Correctly handled
- ✅ Gene clusters are species-specific: Cross-species comparison uses SEED/KEGG annotations, not cluster IDs
- ✅ seedannotation used for functional descriptions (not seedclass)
- ✅ Large table filters: Always filters by orgId when touching genefitness
Notebook organization: NB03 and NB04 follow a clean, consistent structure: markdown header (purpose, inputs, outputs, requirements) → setup → data loading → analysis → QC → visualization → summary. Section headers with numbered markdown cells make the flow easy to follow. NB04 is the densest (25 cells, 8 sections) but remains well-organized with clear progression from overview → core question → stratification → functional enrichment.
Shell script: run_diamond.sh is well-written with set -euo pipefail, proper argument validation with usage message, a cleanup trap for temp files, progress reporting, and caching (skip organisms with existing output). The DIAMOND parameters (--id 90 --max-target-seqs 1) are appropriate for same-species high-identity searches.
src/ scripts: Both run_pipeline.py and extract_essential_genes.py are well-structured, with proper Spark session management via berdl_notebook_utils.setup_spark_session, progress reporting, and caching checks (skip if output file exists). The pipeline design — extract to TSV, then analyze locally — is the correct pattern for BERDL work.
Minor code issues:
1. In NB01 (cell cell-name-match), the NCBI name matching interpolates genus and species directly into SQL LIKE patterns without escaping single quotes. The run_pipeline.py version correctly handles this with replace("'", "''"). The notebook version should do the same for consistency.
2. In NB03, the conservation stacked bar chart (cell cell-qc-conservation) plots core + auxiliary + singleton as additive bars. Since singletons are a subset of auxiliary (documented in docs/pitfalls.md), the chart visually overstates the total. The text output is correct — only the visualization is affected.
3. The NB03 spot check shows MR1:SO_0001 → NO MATCH and MR1:SO_0002 → NO MATCH without explanation. Since MR1 has 96.9% coverage overall, these are likely edge cases worth a brief note.
Findings Assessment
Core result well-supported: The finding that essential genes are modestly enriched in core clusters (median OR=1.56, 18/33 organisms significant after BH-FDR) is convincingly demonstrated through the forest plot, per-organism 2×2 tests, and appropriate multiple testing correction. The honest characterization as "modest" enrichment — noting that most genes in well-characterized bacteria are core regardless of essentiality — shows good scientific judgment.
Functional analysis adds depth: The four-way breakdown (essential-core / essential-auxiliary / essential-unmapped / nonessential) with enzyme classification and SEED/KEGG category analysis goes well beyond the core question:
- Essential-core genes: 41.9% enzymes, 13.0% hypothetical, enriched in Protein Metabolism (+13.7 pp), Cofactors/Vitamins (+6.2 pp)
- Essential-auxiliary genes: 13.4% enzymes, 38.2% hypothetical, top subsystems include ribosomes, DNA replication, type 4 secretion
- Essential-unmapped genes: 58.1% hypothetical, dominated by divergent ribosomal proteins (L34, L36, S11, S12), transposases, DNA-binding proteins
- The depletion of Carbohydrates (-7.9 pp) and Membrane Transport (-4.0 pp) in essential-core genes — "functions that tend to be conditionally important" — is a biologically satisfying interpretation
Limitations comprehensively acknowledged: The README's five-point Limitations section covers essentiality definition bias, small-clade core fraction inflation, E. coli exclusion, single growth condition, and coverage-based organism exclusion. The gene length validation (NB04 Section 1) directly tests the most important potential confound.
Literature context: Five relevant references cited with DOIs and PMIDs. The Rosconi et al. (2022) parallel is particularly apt — their universal essential / core strain-specific / accessory essential categories map directly onto essential-core / essential-auxiliary / essential-unmapped. The Hutchison et al. (2016) quantitative comparison (31% unknown function in minimal genome vs 44.7% hypothetical in essential-unmapped) strengthens the findings.
Stratification analyses: The Spearman correlations between odds ratio and clade size/openness (NB04 Section 4) and the lifestyle stratification (NB04 Section 5) provide important context, addressing whether the enrichment is an artifact of pangenome structure vs a genuine biological pattern.
Number inconsistencies between README and NB04: The README reports some figures that appear to predate the Dyella79 exclusion in NB04:
- README: "34 organisms" → NB04 actual: 33 (after Dyella79 exclusion)
- README: "28,399 putative essential genes" → NB04: 27,693
- README: "153,143 protein-coding genes" → NB04: 148,826
- README: "1,965" essential-unmapped → NB04: 1,259
- README: "18 of 33 organisms" significant → NB04: 18/33 (this one matches)
These are not errors in the analysis — the NB04 code and outputs are internally consistent. The README just needs to be updated to reflect the final 33-organism dataset. The key results (OR=1.56, 18/33 significant, functional breakdown percentages) are correct in both places.
Suggestions
-
[Important] Reconcile README numbers with NB04 actuals: Update the README to consistently use the 33-organism, post-Dyella79 numbers: 148,826 total protein-coding genes, 27,693 essential (18.6%), 1,259 essential-unmapped. The percentages and statistical results in the README already match NB04, so this is primarily a matter of updating the raw counts and the "34 organisms" references.
-
[Important] Add
seed_hierarchy.tsvextraction toextract_essential_genes.py: The SQL query for generatingseed_hierarchy.tsvis documented in a NB04 markdown cell and actually included inextract_essential_genes.py(lines 166–179) — however, it has a caching check that skips if the file exists. This is good. But a reader following the reproduction steps from scratch needs to know this file is generated byextract_essential_genes.pystep 4, not a separate manual step. A note in the README's reproduction section would help. -
[Moderate] Save NB01 and NB02 notebook outputs: These two Spark-dependent notebooks have 0 code cells with saved outputs (vs 15/15 and 18/18 for NB03 and NB04). Since they require BERDL JupyterHub access to re-run, a reader cannot verify organism matching statistics, QC results, or extraction progress without Spark access. Running
jupyter nbconvert --executeand saving the executed notebooks would preserve this narrative context alongside the cached TSV files. -
[Minor] Fix NB03 conservation stacked bar chart: The bar chart in
cell-qc-conservationplots core + auxiliary + singleton as additive bars. Since singletons ⊂ auxiliary (perdocs/pitfalls.md), this overstates the total. Plot as core + (auxiliary − singleton) + singleton, or core + auxiliary with singleton as an overlay/annotation. -
[Minor] Explain MR1 spot-check failures in NB03:
MR1:SO_0001andMR1:SO_0002show "NO MATCH" despite 96.9% overall coverage. A brief note explaining why (e.g., short genes below identity threshold, or non-standard locus tag format for these specific genes) would help readers interpret the QC results. -
[Minor] Escape single quotes in NB01 organism name matching: The notebook version of the NCBI name matching (cell
cell-name-match) interpolates genus/species directly into SQL without escaping. Apply the samereplace("'", "''")fix used insrc/run_pipeline.pyfor consistency and to guard against species names containing apostrophes. -
[Nice-to-have] Add DIAMOND query coverage filter:
run_diamond.shuses--id 90but no--query-coverflag. The high median identity (100.0%) and coverage (94.2%) suggest current results are clean, but adding--query-cover 70would guard against partial-length matches in future runs. -
[Nice-to-have] Clean up empty
data/strain_fastas/directory: This directory exists (10 bytes, created 2026-02-10) but appears to be from an abandoned approach. Either remove it or add a note explaining its purpose.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Conservation Breakdown
Essential Enrichment By Context
Essential Enrichment By Lifestyle
Essential Enzyme Breakdown
Essential Length Validation
Essential Seed Toplevel Heatmap
Essential Vs Core Forest Plot
Identity Distributions