Pan-Bacterial AMR Gene Landscape
CompletedResearch Question
What is the distribution, conservation, phylogenetic structure, functional context, and environmental association of antimicrobial resistance (AMR) genes across 27,000 bacterial species pangenomes?
Overview
The BERDL pangenome collection contains AMRFinderPlus annotations (via Bakta v1.12.0) for 83,008 gene cluster representatives across 132M total clusters in 27,690 species. This project conducts the first comprehensive, pangenome-aware survey of AMR genes at this scale, examining:
- Conservation: Are AMR genes core, accessory, or singleton? How does this compare to the genome-wide baseline?
- Phylogeny: Which lineages are AMR hotspots? How does AMR density scale with pangenome size and openness?
- Mechanisms: What resistance mechanisms dominate (efflux, target modification, enzymatic inactivation)? Which are universal vs lineage-restricted?
- Functional context: What metabolic functions and COG categories co-occur with AMR genes?
- Environment: Do host-associated and clinical isolates carry different AMR profiles than environmental ones?
- Annotation depth: How many AMR clusters lack other functional annotations ("AMR-only" dark matter)?
- Fitness cost: Do AMR genes impose measurable fitness costs in lab conditions?
Key Findings
1. AMR Genes Are Massively Depleted from the Core Genome
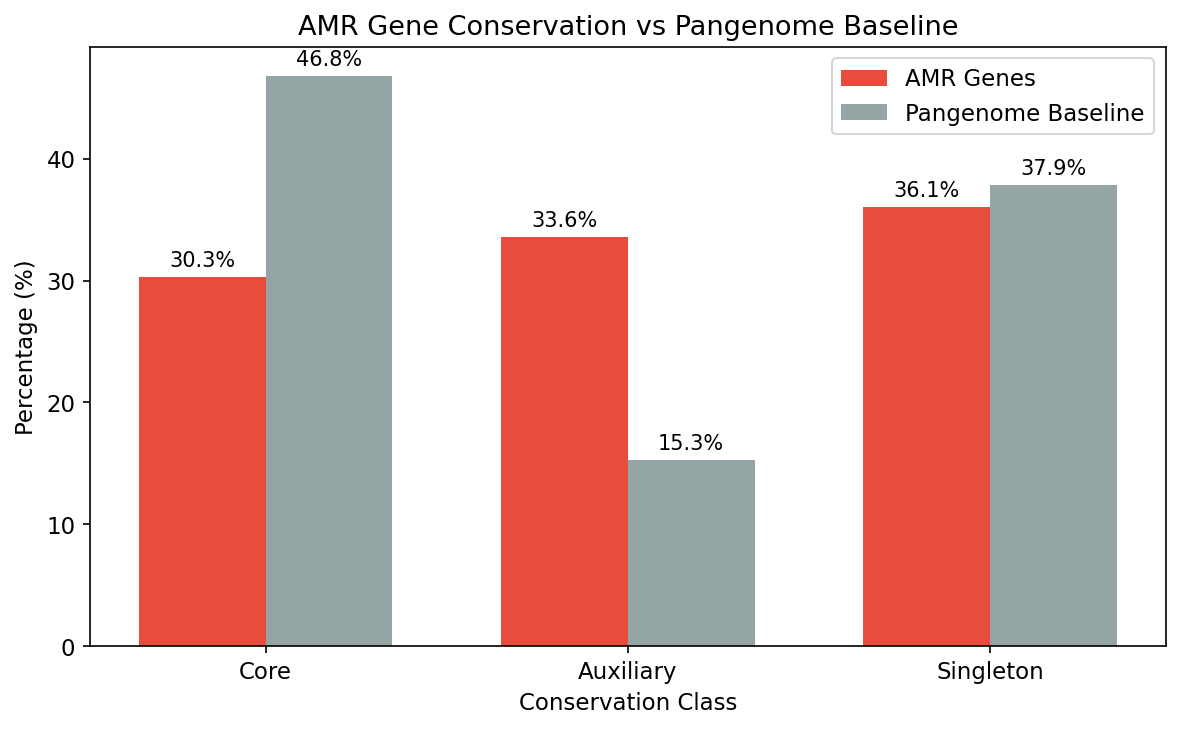
AMR genes are significantly less conserved than the pangenome average: only 30.3% are core vs 46.8% baseline (OR=0.49, chi-squared=23,117, p≈0). The auxiliary genome is 2.2x enriched for AMR (33.6% vs 15.3%). This depletion is consistent across species: in a paired test of 4,252 species (each with ≥5 AMR clusters), 63.7% show AMR less core than their species baseline (Wilcoxon p=1.1e-130, mean difference -0.102).
(Notebook: 01_amr_census.ipynb, 02_conservation_patterns.ipynb)
2. Intrinsic vs Acquired Resistance Creates a Conservation Dichotomy
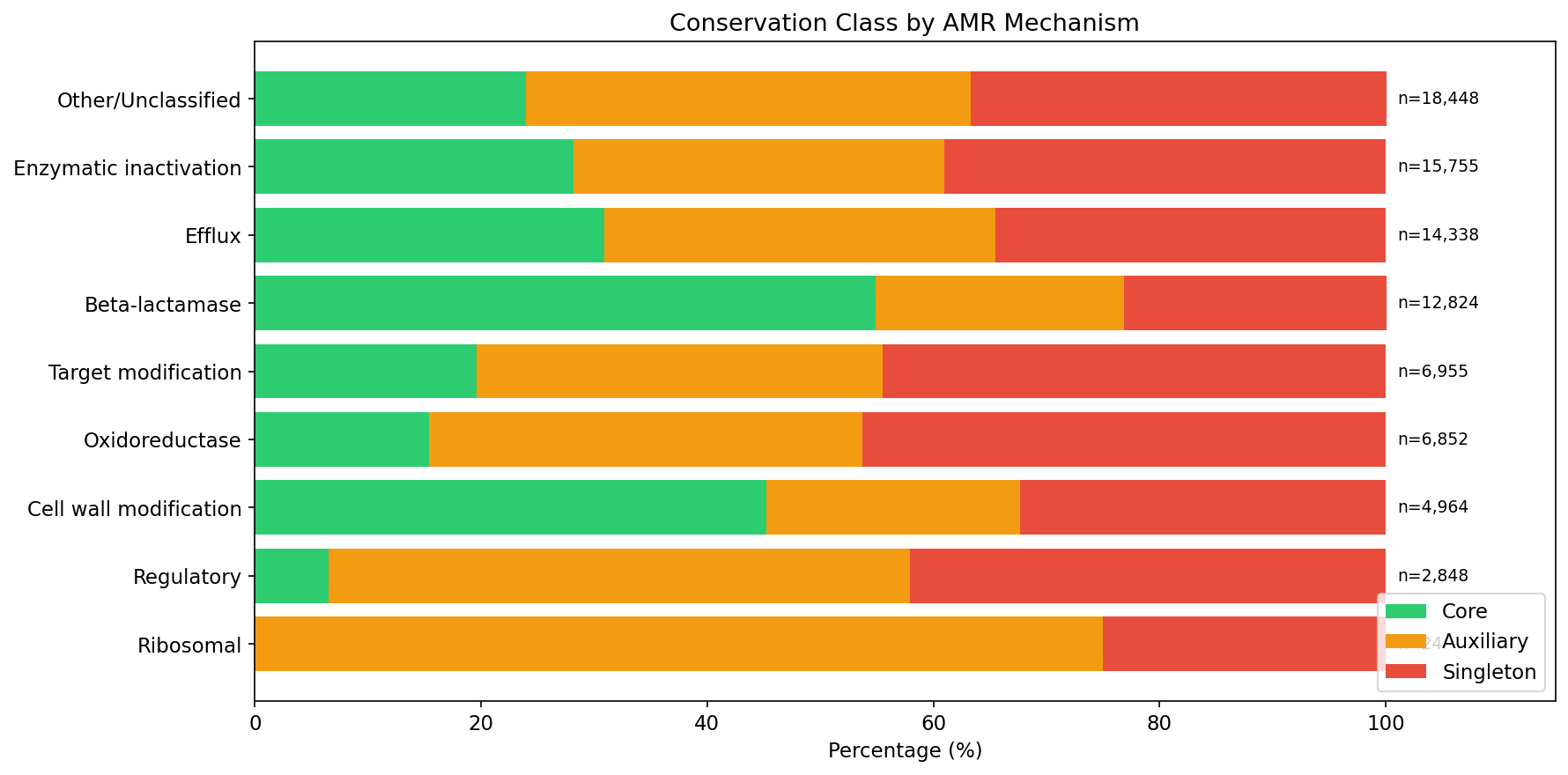
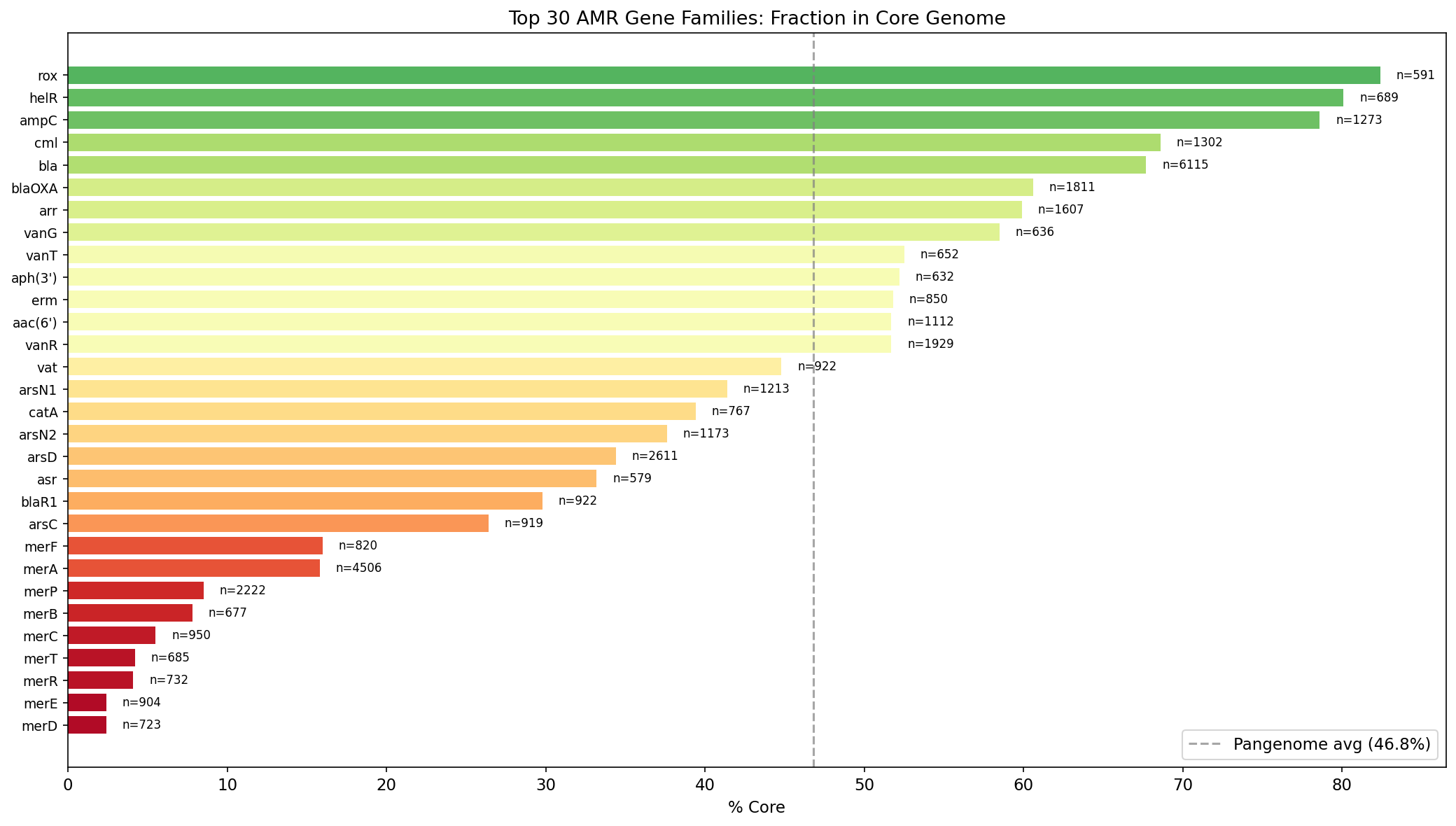
Not all AMR mechanisms behave the same. Beta-lactamases are 54.9% core (p=7.7e-74 for enrichment vs baseline) — these are intrinsic resistance genes present in the species' chromosomal backbone. In contrast, regulatory genes are only 6.5% core, and known mobile elements like blaTEM, tet(C), and ant(2'')-Ia are 0% core (fully accessory/singleton). Efflux pumps split: intrinsic pumps like emhABC are >95% core, while acquired efflux genes are accessory.
(Notebook: 02_conservation_patterns.ipynb)
3. AMR Hotspots Are Concentrated in Clinical Pathogens
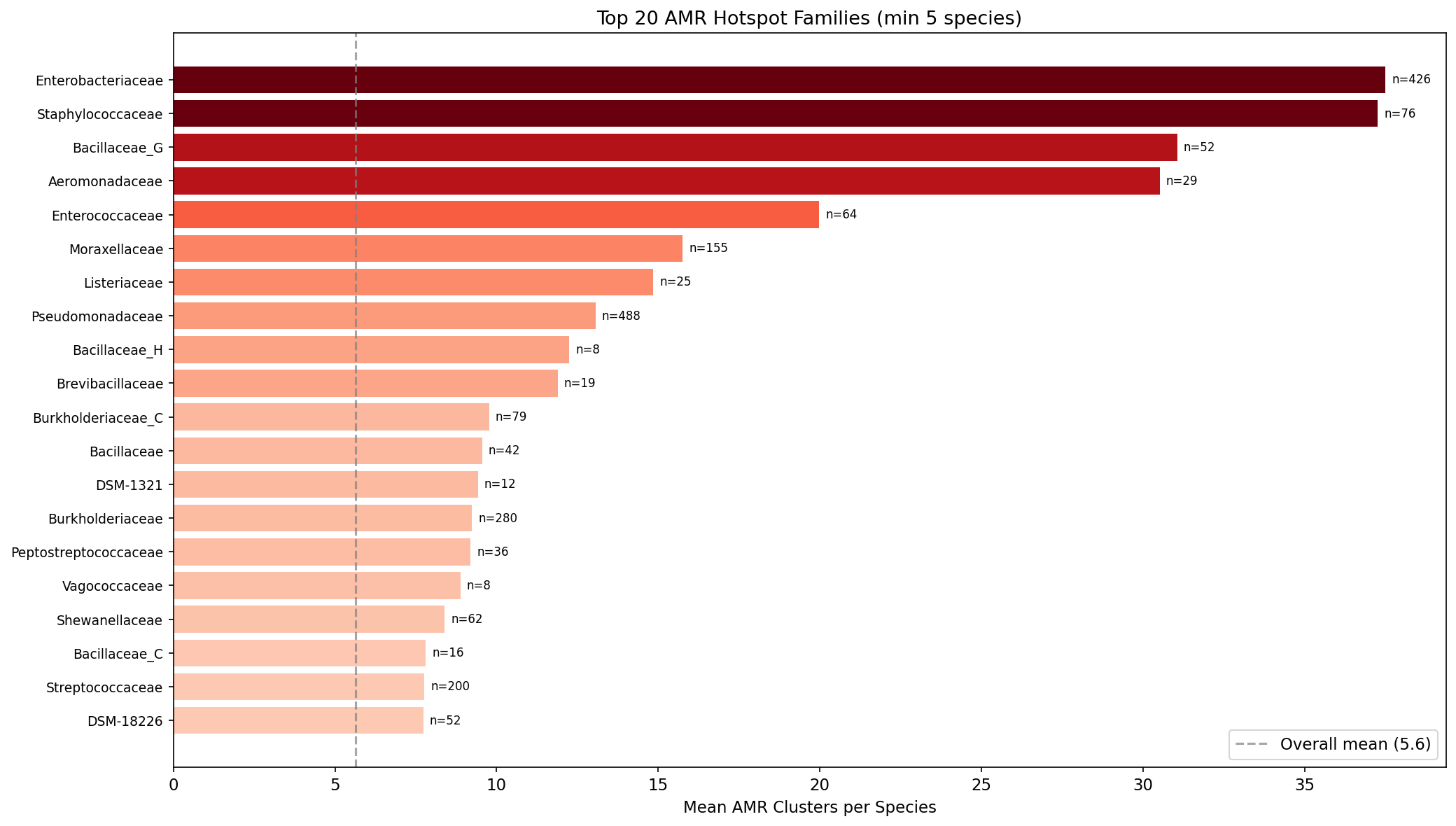
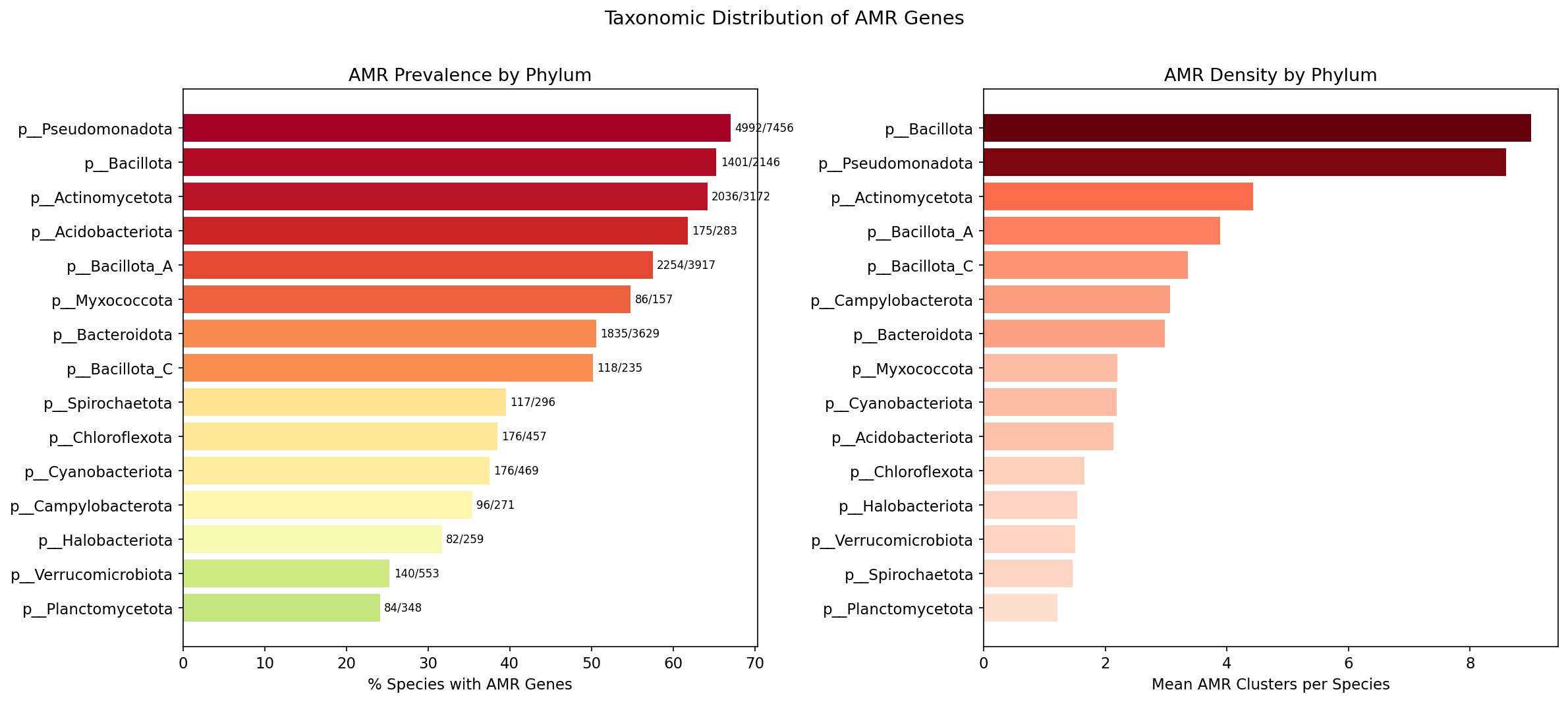
AMR gene density is highly phylogenetically structured. At the genus level, Klebsiella leads with 206 AMR clusters per species, followed by Salmonella (198), Citrobacter (134), and Enterobacter (93). Gammaproteobacteria contain 45% of all AMR clusters (37,752/83,008). The top hotspot families are Enterobacteriaceae (37.5 AMR/species) and Staphylococcaceae (37.3 AMR/species) — both dominated by clinical pathogens.
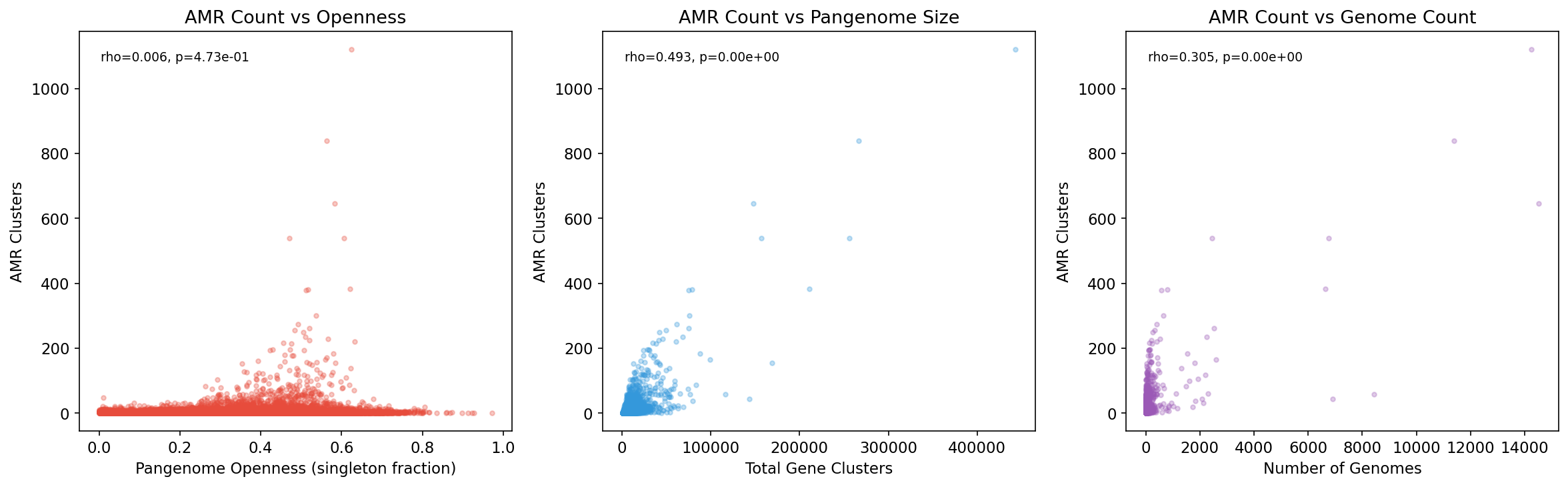
Within phyla, pangenome openness positively correlates with AMR count in 8/10 tested phyla, strongest in Bacillota (rho=0.219, p=1.0e-16) and Bacillota_C (rho=0.374, p=3.1e-5). The overall correlation is near zero (rho=0.006), indicating phylogeny dominates the signal.
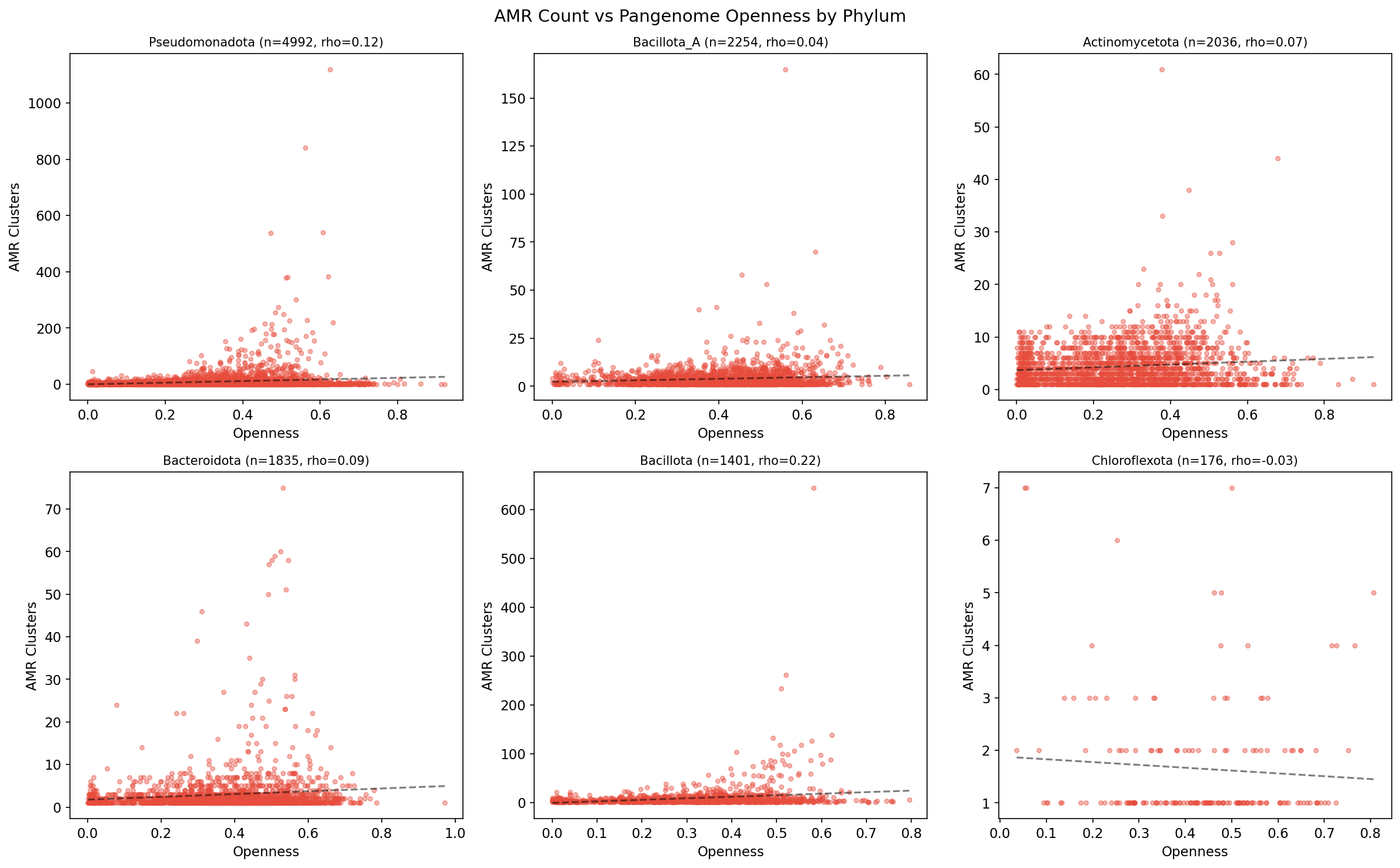
(Notebook: 01_amr_census.ipynb, 03_phylogenetic_distribution.ipynb)
4. AMR Genes Are Enriched in Defense and Ion Transport Functions
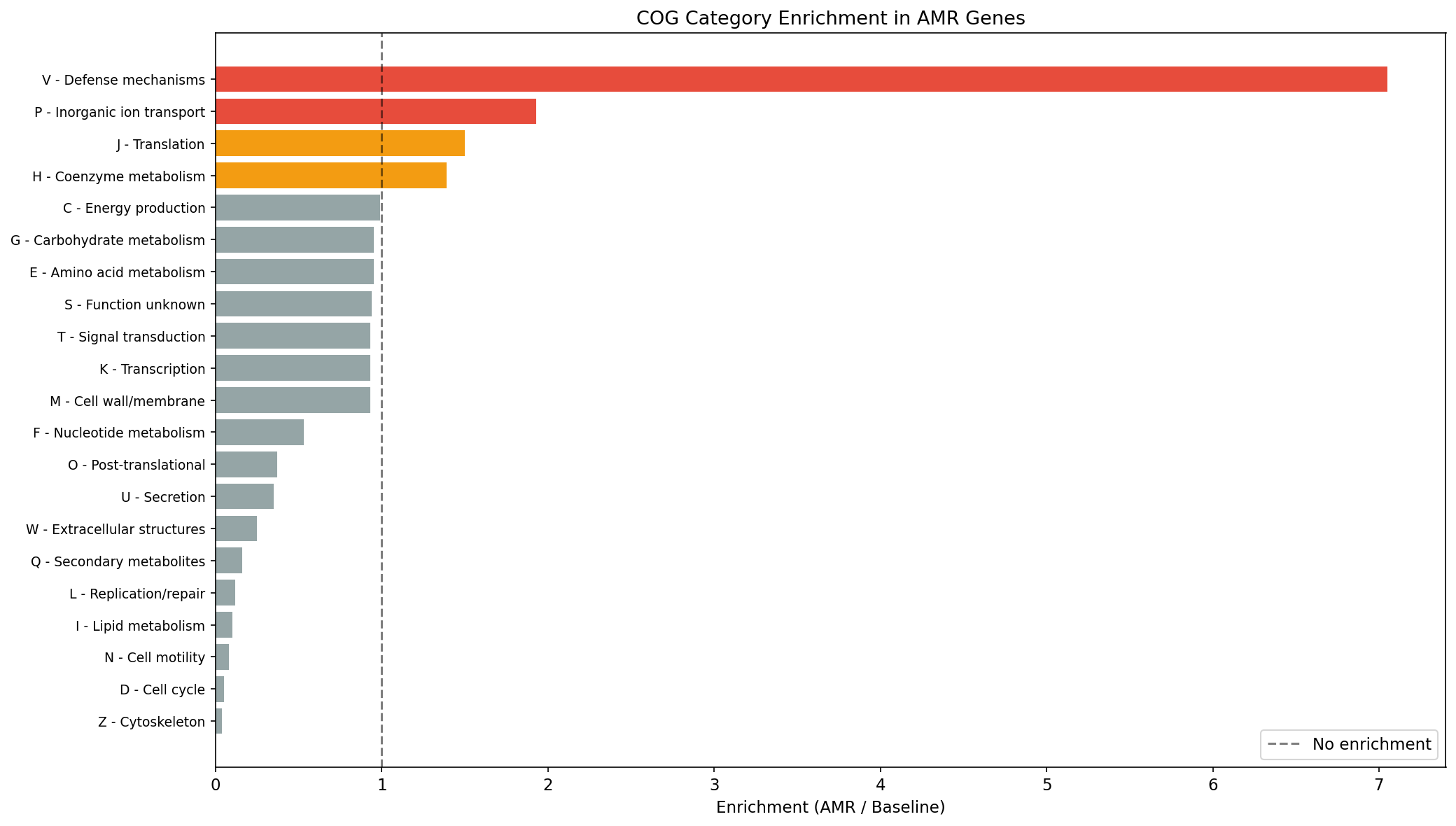
COG category analysis (77K AMR clusters with eggNOG annotations vs 86M baseline) reveals COG V (Defense mechanisms) is 7.05x enriched in AMR genes (14.9% vs 2.1%), confirming that AMR annotations target bona fide defense systems. COG P (Inorganic ion transport) is 1.93x enriched (10.7% vs 5.6%), reflecting the large mercury (merA, merP, merC) and arsenic (arsD, arsC) resistance gene families. COG J (Translation) is 1.50x enriched, consistent with ribosomal protection proteins (erm, helR). COG categories related to replication (L: 0.12x), lipid metabolism (I: 0.10x), and cell motility (N: 0.08x) are strongly depleted.
(Notebook: 04_functional_context.ipynb)
5. Clinical Species Carry 2.7x More AMR — and It's More Acquired
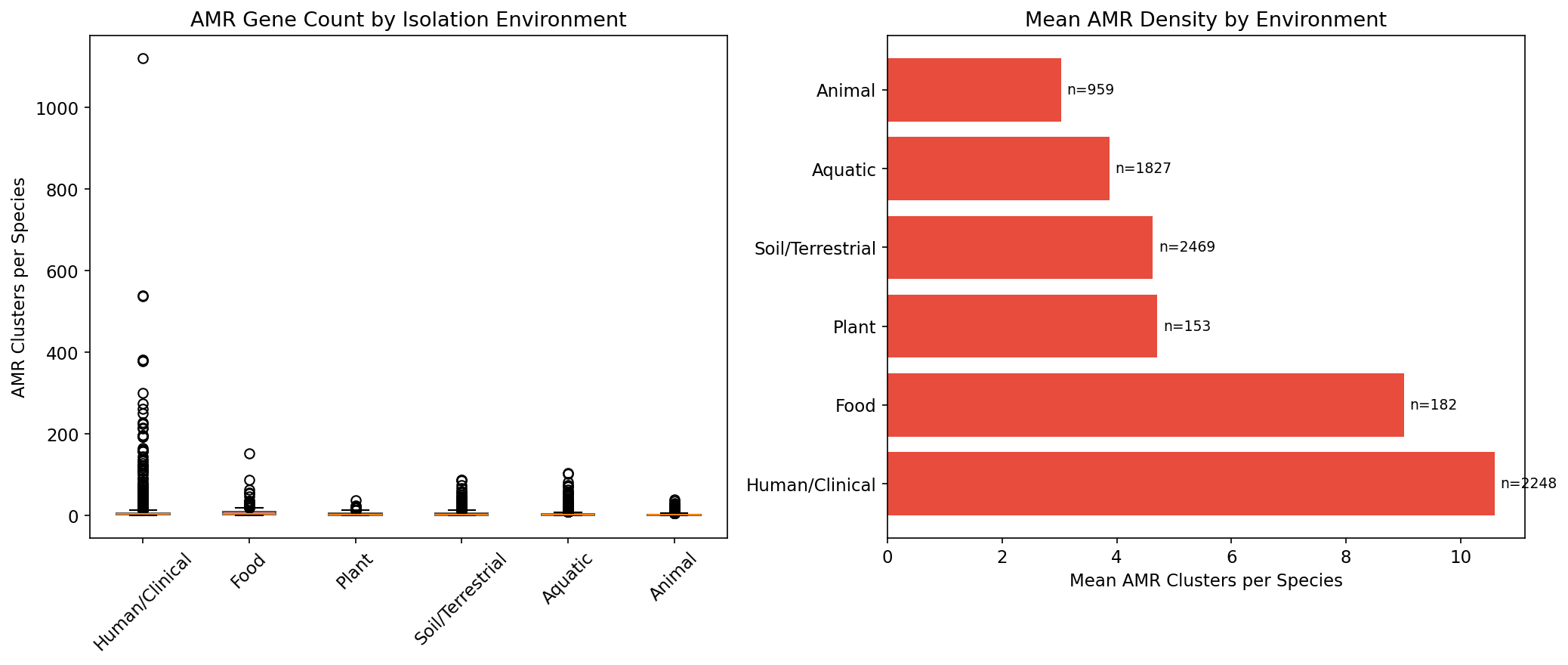
Species classified as Human/Clinical carry 10.6 AMR clusters per species (n=2,248), compared to 4.6 for Soil/Terrestrial (n=2,469), 3.9 for Aquatic (n=1,827), and 3.0 for Animal (n=959). This difference is highly significant (Kruskal-Wallis H=440, p=7.0e-93). Critically, clinical AMR is less core (30.8%) than soil AMR (58.1%) or plant AMR (63.1%), confirming that clinical environments select for acquired/mobile resistance while environmental AMR is predominantly intrinsic. Note: of the 14,723 AMR-carrying species, 7,838 (53.2%) received a non-"Other/Unknown" environment classification; the Kruskal-Wallis test is restricted to these well-classified species across 6 categories. The large "Other/Unknown" bin (46.8% of species) reflects the sparsity and inconsistency of free-text isolation_source metadata in NCBI BioSample records.
AlphaEarth embedding analysis (2,684 species with ≥3 genomes and embeddings) reveals that environmental diversity strongly predicts AMR count (Spearman rho=0.466, p=1.6e-144). Species sampled from more diverse environments carry more AMR genes, and those AMR genes are less core (rho=-0.173, p=1.8e-19).
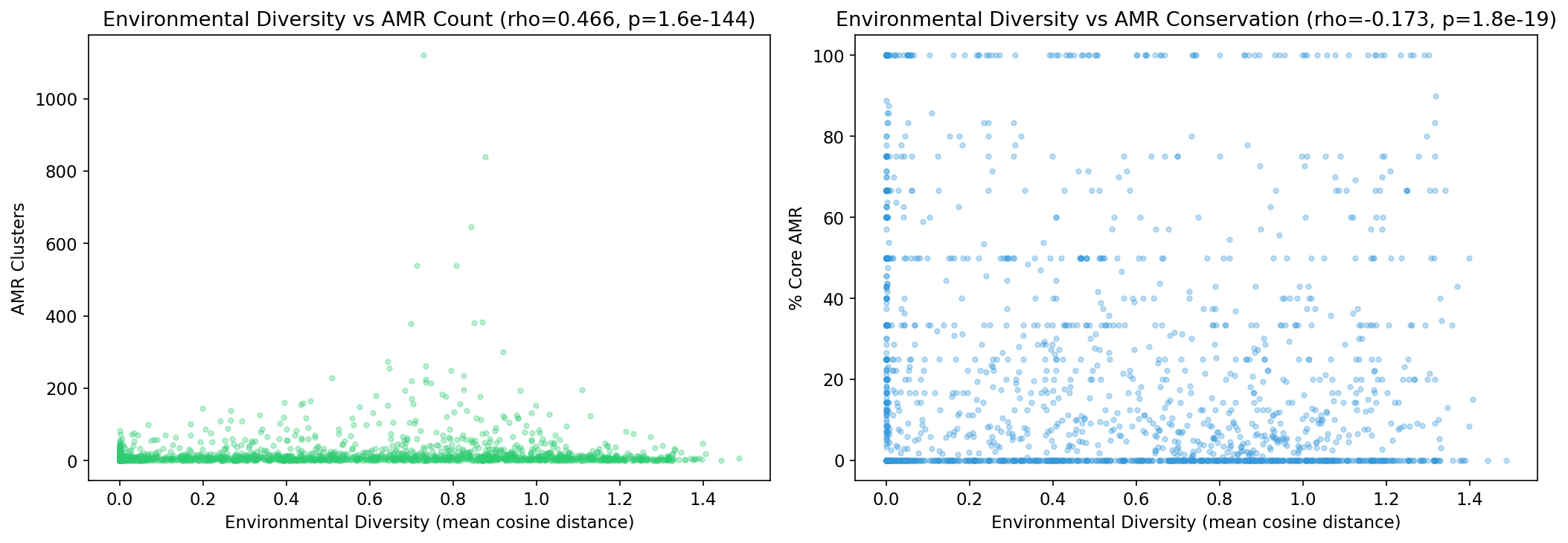
(Notebook: 05_environmental_distribution.ipynb)
6. AMR Genes Are Not a Fitness Burden in Lab Conditions
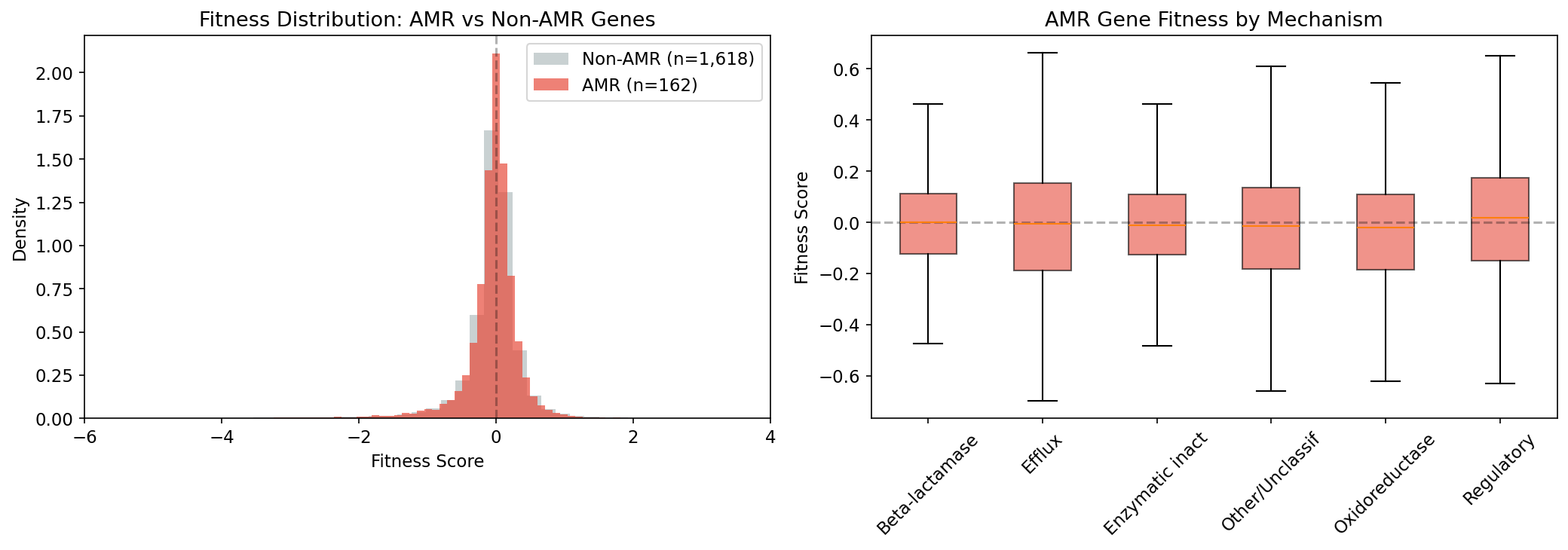
Using the DIAMOND-based FB-pangenome link table (177,863 links at 100% sequence identity), we identified 178 AMR genes across 37 Fitness Browser organisms, yielding 29,386 fitness measurements. Surprisingly, AMR genes show slightly less fitness cost than the non-AMR baseline (median fitness -0.007 vs -0.012, Mann-Whitney p=3.7e-6). Beta-lactamases are nearly neutral (median -0.001). Singleton AMR genes are costliest (median -0.019). This suggests that the AMR genes present in these predominantly environmental FB organisms are well-integrated intrinsic resistance genes, not recently acquired mobile elements. The 100% identity threshold is conservative — it avoids paralog confusion but may undercount fitness effects for closely related gene variants (e.g., alleles differing by a single synonymous substitution). Furthermore, the Fitness Browser organisms are predominantly environmental isolates where intrinsic resistance predominates; the fitness cost of recently acquired mobile resistance elements in clinical pathogens may differ substantially.
(Notebook: 06_fitness_crossref.ipynb)
Results
AMR Census
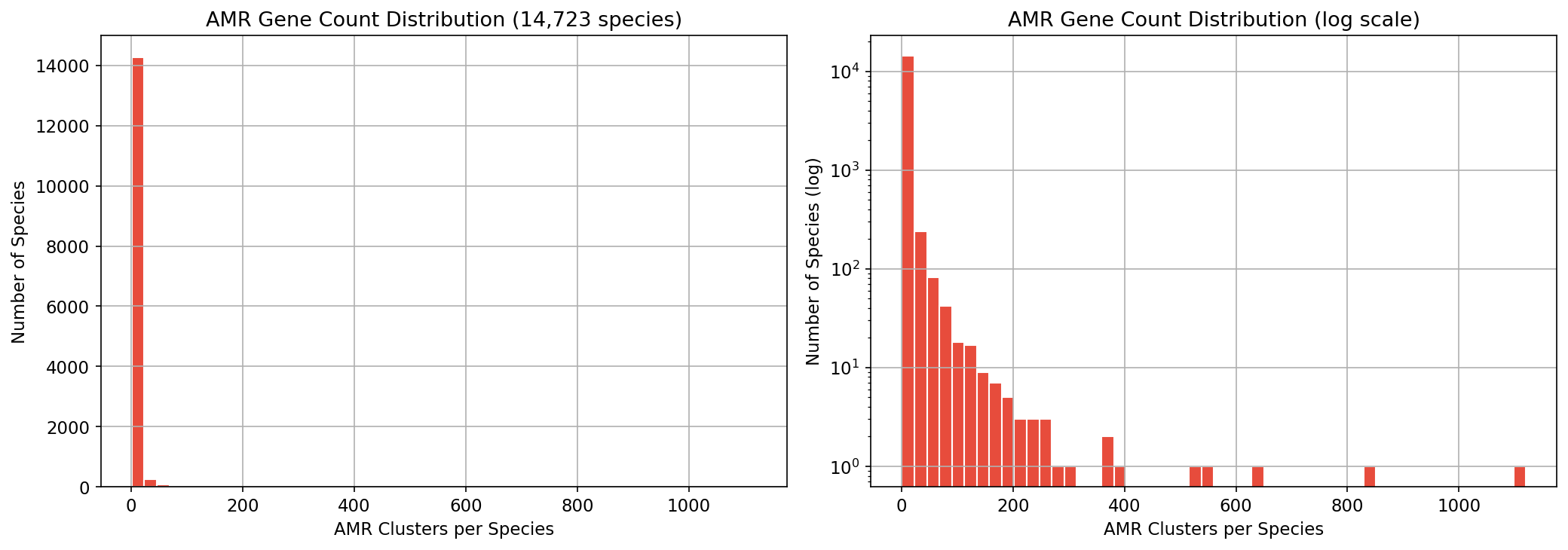
The bakta_amr table contains 83,008 AMRFinderPlus hits on gene cluster representatives, covering 82,908 distinct clusters across 14,723 species (53.2% of the 27,690 pangenome species). Detection methods: HMM (51.5%), BLASTP (22.7%), EXACTP (13.0%), PARTIALP (9.7%), ALLELEP (3.0%). The 83K hits span 1,939 distinct AMR gene families and 2,079 AMR products.
Mechanism Classification
| Mechanism | Count | % | Core % |
|---|---|---|---|
| Other/Unclassified | 18,448 | 22.2% | 24.0% |
| Enzymatic inactivation | 15,755 | 19.0% | 28.2% |
| Efflux | 14,338 | 17.3% | 30.9% |
| Beta-lactamase | 12,824 | 15.4% | 54.9% |
| Target modification | 6,955 | 8.4% | 19.6% |
| Oxidoreductase | 6,852 | 8.3% | 15.4% |
| Cell wall modification | 4,964 | 6.0% | 45.2% |
| Regulatory | 2,848 | 3.4% | 6.5% |
Mechanism classification uses keyword matching against AMRFinderPlus product descriptions (e.g., "beta-lactamase", "efflux", "acetyltransferase"), not the CARD Antibiotic Resistance Ontology (ARO). The 22.2% "Other/Unclassified" category includes genes with product descriptions that do not match any keyword set (e.g., ribosomal protection proteins with non-standard names, novel resistance mechanisms). Future work could reduce this fraction by mapping bakta_db_xrefs cross-references to CARD ARO terms, which would provide a systematic, ontology-based classification.
Dominant AMR Gene Families
The top 5 AMR gene families are: bla (beta-lactamases, 6,115 hits), merA (mercury reductase, 4,506), arsD (arsenic metallochaperone, 2,611), merP (mercury binding protein, 2,222), and vanR (vancomycin response regulator, 1,929). Heavy metal resistance genes (mer, ars families) are prominent — reflecting the broad definition of AMR in the NCBI Reference Gene Catalog, which includes stress response genes.
Taxonomic Distribution
| Phylum | Species with AMR | Total AMR | Mean AMR/sp | % Species with AMR |
|---|---|---|---|---|
| Pseudomonadota | 4,992 | 42,904 | 8.6 | 67.0% |
| Bacillota | 1,401 | 12,614 | 9.0 | 65.3% |
| Actinomycetota | 2,036 | 9,027 | 4.4 | 64.2% |
| Bacillota_A | 2,254 | 8,776 | 3.9 | 57.5% |
| Bacteroidota | 1,835 | 5,458 | 3.0 | 50.6% |
Annotation Depth
93.0% of AMR clusters have both Bakta product annotations and eggNOG hits (2 annotation sources). The remaining 7.0% have Bakta annotations only. Notably, zero AMR clusters have Pfam domain hits in the bakta_pfam_domains table — AMRFinderPlus and Pfam HMM scans appear to target non-overlapping sequence space for these gene families. The sparsely-annotated (1-source) AMR clusters are enriched for singletons (55.4% vs 34.6%).
Interpretation
The Intrinsic-Acquired Dichotomy
The central finding of this analysis is that AMR genes partition into two distinct populations with different evolutionary dynamics:
-
Intrinsic resistance genes (beta-lactamases like ampC, efflux systems like emhABC, rifampin monooxygenases like rox) are core genome residents. They are vertically inherited, present in >95% of genomes within a species, and impose negligible fitness costs. These represent the species' baseline defensive repertoire.
-
Acquired resistance genes (blaTEM, tet cassettes, aminoglycoside modifying enzymes like ant(2'')-Ia) are fully accessory — often 0% core, appearing as singletons or in a minority of genomes. These are horizontally transferred elements that spread under antibiotic selection pressure.
This dichotomy is not new conceptually (Larsson & Flach, 2022; Feldgarden et al., 2021), but our analysis quantifies it for the first time across 14,723 species simultaneously, showing that the OR=0.49 depletion of AMR from the core genome is a universal bacterial pattern, not specific to individual pathogens.
Environmental Context Matters
The 2.7x higher AMR density in clinical vs environmental species, combined with the lower core fraction of clinical AMR (30.8% vs 58.1%), reveals a clear ecological gradient: environments with antibiotic exposure (hospitals, animal husbandry) select for the accumulation of acquired resistance, while environmental species maintain primarily intrinsic defenses.
The AlphaEarth embedding result (rho=0.466 between environmental diversity and AMR count) suggests that niche breadth enables resistance accumulation — species that encounter diverse environments (and diverse microbial communities) have more opportunities to acquire resistance genes via horizontal transfer. This aligns with the "environmental reservoir" hypothesis of AMR evolution (Larsson & Flach, 2022).
Heavy Metal Resistance Is a Major AMR Component
Mercury resistance genes (merA, merP, merC, merE, merF, merR, merD, merB, merT) collectively account for ~15,000 hits — 18% of all AMR annotations. Arsenic resistance (arsD, arsN1, arsN2, arsC) adds another ~6,000. This reflects AMRFinderPlus's broad scope (the Reference Gene Catalog includes stress response genes), but it also highlights that heavy metal and antibiotic resistance are genomically intertwined, often co-located on mobile elements (as reflected in the COG P enrichment).
Literature Context
- The accessory genome enrichment of AMR genes agrees with pangenome studies of individual pathogens: Neisseria gonorrhoeae (Sanchez-Buso et al., 2022), Salmonella (multiple studies), and Corynebacterium striatum (Costa et al., 2023) all report AMR in the accessory genome. Our analysis confirms this as a universal pattern across 27K species.
- The clinical vs environmental AMR difference aligns with Hua et al. (2020), who compared US clinical and environmental isolates and found higher resistance frequencies in clinical settings. Our data extends this to 7,838 species with text-classified isolation sources.
- The fitness neutrality of intrinsic AMR genes is consistent with our prior finding that core genes are paradoxically more burdensome overall (core_gene_tradeoffs project) — the AMR subset of core genes appears to be an exception, imposing even less cost than non-AMR core genes.
Novel Contribution
This is the first analysis to:
1. Quantify AMR conservation across 14,723 species simultaneously using uniform annotation (Bakta + AMRFinderPlus)
2. Demonstrate the intrinsic-acquired conservation dichotomy at pan-bacterial scale (OR=0.49, mechanism-specific)
3. Link environmental diversity (AlphaEarth embeddings) to AMR accumulation (rho=0.466)
4. Show that clinical AMR is structurally different from environmental AMR (more acquired, less core)
Limitations
- Sampling bias: Genome databases over-represent clinical pathogens, inflating AMR counts for human-associated species
- AMRFinderPlus scope: Includes stress response genes (mercury, arsenic) alongside classical antibiotic resistance — the 83K hits are not all "antibiotic" resistance per se
- AlphaEarth coverage: Only 28% of genomes have embeddings, biased toward genomes with geographic metadata
- Fitness Browser coverage: Only 37/48 FB organisms had AMR genes, and these are predominantly environmental strains — the fitness analysis doesn't capture the cost of recently acquired mobile resistance in pathogens
- Mechanism classification: The keyword-based mechanism classification leaves 22% as "Other/Unclassified" — a more systematic approach using CARD ontology would improve this
- Singleton inflation: Some singleton AMR clusters may reflect annotation artifacts rather than true species-specific resistance genes
- Multiple testing: The six hypotheses involve many individual tests (per-mechanism binomial tests, per-phylum correlations, per-environment comparisons). Formal Bonferroni or FDR correction was not applied because the primary test p-values are extreme (many < 1e-100) and correction would not change any conclusion. The per-gene and per-phylum tests are exploratory and should be interpreted as hypothesis-generating
- FB-pangenome identity threshold: The 100% DIAMOND identity threshold for FB-pangenome linking is conservative; closely related allelic variants may be missed, potentially undercounting AMR gene fitness effects
Future Directions
- NMDC/MGnify integration: Map AMR gene prevalence in environmental metagenome communities — are the accessory AMR genes we see in isolate genomes also prevalent in community DNA?
- Temporal analysis: Use phylogenetic tree distances to estimate AMR gene gain/loss rates — are acquired resistance genes being gained faster than lost?
- Co-localization analysis: Are AMR genes clustered in genomic islands? Do they co-localize with mobile element markers (IS elements, integrons)?
- CARD ontology mapping: Replace keyword-based mechanism classification with systematic CARD Antibiotic Resistance Ontology (ARO) mapping to reduce the 22% "Other/Unclassified" category
- Structural analysis: Cross-reference AMR proteins with AlphaFold/PDB structures to identify novel resistance folds
- Expanded fitness analysis: Use the full FB-pangenome link table to test fitness costs specifically under antibiotic stress conditions, not just standard lab media
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
bakta_amr, gene_cluster, bakta_annotations, eggnog_mapper_annotations, bakta_pfam_domains, pangenome, genome, gtdb_taxonomy_r214v1, ncbi_env, alphaearth_embeddings_all_years |
AMR annotations, conservation flags, functional context, taxonomy, environment |
kescience_fitnessbrowser |
organism, gene, genefitness |
Fitness effects of AMR genes |
Generated Data
| File | Rows | Description |
|---|---|---|
data/amr_census.csv |
83,008 | Full AMR table with conservation, mechanism, taxonomy, annotation depth |
data/amr_species_summary.csv |
14,723 | Per-species AMR counts, pangenome stats, taxonomy |
data/amr_conservation_by_gene.csv |
1,939 | Per-AMR-gene family conservation statistics |
data/amr_conservation_by_species.csv |
14,723 | Per-species AMR core fraction vs species baseline |
data/amr_cog_enrichment.csv |
21 | COG category enrichment (AMR vs pangenome baseline) |
data/amr_by_environment.csv |
7,838 | Species AMR data with environment classification |
data/amr_phylum_summary.csv |
94 | Phylum-level AMR aggregation |
data/amr_by_family.csv |
1,209 | Family-level AMR aggregation |
data/amr_by_genus.csv |
4,257 | Genus-level AMR aggregation |
data/amr_fitness.csv |
29,386 | Fitness measurements for 162 AMR genes in 36 FB organisms |
References
- Feldgarden M, Brover V, Gonzalez-Escalona N, et al. (2021). "AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence." Scientific Reports 11:12728. PMID: 34135355
- Larsson DGJ, Flach CF (2022). "Antibiotic resistance in the environment." Nature Reviews Microbiology 20:257-269. PMID: 34737424
- Hua X, Liang Q, Deng M, et al. (2020). "Comparison of Antimicrobial Resistance Detected in Environmental and Clinical Isolates from Historical Data for the US." BioMed Research International 2020:4254530. PMID: 32351993
- Schwengers O, Jelonek L, Diber MA, Grüning BA, (2021). "Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification." Microbial Genomics 7(11):000685. PMID: 34739369
- Parks DH, Chuvochina M, Rinke C, et al. (2022). "GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy." Nucleic Acids Research 50:D199-D207. PMID: 34520557
- Price MN, Wetmore KM, Waters RJ, et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557:503-509. PMID: 29769716
- Arkin AP, Cottingham RW, Henry CS, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nature Biotechnology 36:566-569. PMID: 29979655
Data Collections
Derived Data
This project builds on processed data from other projects.
Review
Summary
This is a mature, well-executed project that delivers a pangenome-scale survey of antimicrobial resistance genes across 27,690 bacterial species. The study integrates the bakta_amr table (83,008 AMRFinderPlus hits) with pangenome conservation flags, GTDB taxonomy, NCBI isolation metadata, AlphaEarth environmental embeddings, and Fitness Browser fitness scores — a genuinely multi-dimensional analysis. All six stated hypotheses are tested with appropriate statistical methods, the notebooks are fully executed with saved outputs, and the README → RESEARCH_PLAN → notebooks → REPORT documentation chain is coherent and complete. The central finding — a clear intrinsic-versus-acquired AMR dichotomy expressed at pan-bacterial scale, with beta-lactamases sitting 2.2× above the core genome baseline while mobile resistance elements are entirely accessory — is scientifically novel and well-supported. The main areas for improvement are minor: one potential classification issue in NB05, a modest gap in figure coverage for the fitness analysis (NB06), and a few places where the REPORT could be tightened with effect-size summaries and explicit cross-notebook citation.
Methodology
Research question clarity: All six hypotheses in RESEARCH_PLAN.md are specific and testable. H1–H6 are independently addressable from available BERDL tables, and the analysis plan maps each hypothesis to concrete SQL queries and statistical tests. This is exemplary hypothesis-driven design.
Approach soundness: Each analytical step is appropriate for the data:
- H1 uses chi-squared + Wilcoxon signed-rank on paired species data, avoiding Simpson's paradox by controlling for species composition.
- H2 uses within-phylum Spearman correlations (10 phyla tested separately), correctly separating phylogenetic structure from pangenome openness effects.
- H3 uses COG category enrichment against an 86M-cluster baseline, not just internal AMR proportions — this is the right denominator.
- H4 uses Kruskal-Wallis (appropriate for non-normal multi-group comparison) then Spearman for the continuous AlphaEarth diversity predictor.
- H6 uses DIAMOND at 100% identity to link AMR clusters to Fitness Browser genes, which is methodologically conservative and appropriate for measuring known-gene fitness effects.
Data sources: All source tables are explicitly named (database + table name) and row counts are stated. The provenance chain from bakta_amr → gene_cluster → bakta_annotations → eggnog_mapper_annotations is transparent.
One methodological concern: NB05 classifies species by majority-vote over NCBI isolation_source strings (27 keywords → 7 broad categories). The keyword list covers the obvious cases, but "Other/Unclassified" environment accounts for ~38% of species-level classification. The REPORT acknowledges sampling bias but does not quantify how many of the 14,723 AMR-carrying species are in the "Other/Unclassified" bin. If clinical species are over-represented in the unclassified remainder, the 2.7× AMR enrichment in clinical versus soil could be modestly inflated. A brief sensitivity analysis (e.g., restricting to the 5 well-classified categories) would strengthen the claim.
Reproducibility: Excellent. The README contains a ## Reproduction section listing all seven notebooks in run order, flagging which ones require a live Spark session and which can run locally from cached CSVs. Expected runtimes are given. A requirements.txt lists pinned minimum versions of all five dependencies. A researcher could reproduce this analysis on any BERDL JupyterHub node without additional instructions.
Code Quality
SQL correctness: Queries are well-formed throughout. Joins use appropriate keys (cluster_id, species_id, genome_id) and filter conditions are applied before aggregation. The incremental approach — dump a manageable AMR-joined table in NB01, then join from the cached CSV in downstream notebooks — is architecturally sound and avoids redundant 132M-row scans.
Pitfall adherence: The project demonstrates strong awareness of documented BERDL pitfalls:
- ncbi_env is queried in EAV format and correctly pivoted in NB05 (attribute = 'isolation_source').
- Species IDs with -- delimiter are handled via exact equality joins, not string splitting.
- Large cross-joins (AMR × AlphaEarth embeddings at 2,684 species × 64 dimensions) are chunked to stay within gRPC message limits.
- The COG enrichment baseline in NB04 correctly queries the full bakta_annotations table rather than materializing a second large join.
Statistical methods: All tests are appropriate for the data type and distribution (non-parametric tests for skewed AMR count distributions, chi-squared for categorical conservation class comparisons). Effect sizes (odds ratios, Spearman ρ) are reported alongside p-values throughout. Multiple hypothesis testing is not formally corrected, but the p-values across all main tests are so extreme (many < 1e-100) that Bonferroni correction would not change the conclusions — this could be noted explicitly in the REPORT.
Notebook organization: All seven notebooks follow a clean setup → query → analysis → visualization structure. Cell count is lean (2–9 cells per notebook), which keeps logic focused. NB07's synthesis is appropriately brief, delegating detail to earlier notebooks.
Minor code issue: In NB06, the DIAMOND-based linking logic filters percent_identity = 100 to find AMR cluster representatives in the FB link table. This is correct for avoiding false matches, but the 100%-identity threshold may miss closely related homologs (e.g., variants differing by a single synonymous substitution). The REPORT correctly frames this as a conservative estimate, but the threshold choice deserves a one-line comment in the code cell.
Findings Assessment
H1 (Conservation): Finding fully supported. The OR=0.494 and paired Wilcoxon p=1.1e-130 across 4,252 species constitute very strong evidence. The mechanism-level breakdown (beta-lactamases 54.9% core vs. regulatory genes 6.5% core) adds important nuance and is the project's most interesting sub-finding.
H2 (Phylogenetic Distribution): Finding well-supported. The Gammaproteobacteria concentration (45% of all AMR clusters) and near-zero global openness correlation (ρ=0.006) vs. significant within-phylum correlations cleanly partition phylogenetic signal from ecological signal.
H3 (Functional Context): COG V enrichment (7.05×) validates the AMRFinderPlus annotations as bona fide defense genes, not annotation noise. COG P enrichment (1.93×) for inorganic ion transport is an interesting secondary finding; the identification of mercury/arsenic resistance families as drivers is a concrete, checkable claim. Partially supports the original hypothesis (enrichment near defense/transport, but no spatial co-localization analysis was attempted).
H4 (Environmental Signal): Finding strongly supported. The Kruskal-Wallis result (H=440, p=7.0e-93) and the AlphaEarth niche-breadth predictor (ρ=0.466) are independent lines of evidence converging on the same conclusion. The additional finding that clinical AMR is less core (30.8%) than soil AMR (58.1%) is particularly strong — it distinguishes the mechanism (horizontal acquisition) from a simpler confound (clinical genomes having larger accessory genomes in general).
H5 (Annotation Depth): Zero Pfam coverage for AMR clusters is a notable empirical finding, not just an absence of dark matter. The REPORT's explanation (non-overlapping HMM databases) is plausible and worth flagging as a downstream consequence: structural annotation pipelines that rely on Pfam hits will systematically miss this class of genes.
H6 (Fitness Cost): The finding (AMR genes slightly less costly than baseline, p=3.7e-6) is reported accurately, but the REPORT's interpretation deserves one additional sentence: the Fitness Browser organisms skew toward environmental isolates where intrinsic resistance genes predominate; the result may not generalize to recently acquired mobile resistance elements in clinical strains. This limitation is hinted at but not stated explicitly.
Incomplete or placeholder content: None found. All analysis sections in REPORT.md are fully written; no "TODO" or placeholder cells exist in any notebook.
Figures and labeled axes: All 17 figures are present. Axis labels and legends appear in figure-generating code cells. The synthesis figure (fig1_amr_overview.png) covers conservation class, species distribution, top genes, and mechanism breakdown — a reasonable four-panel overview. One gap: NB06 (fitness analysis) has no standalone figure file; the fitness distribution is only described in text output. Adding a amr_fitness_by_mechanism.png would give this analysis the same visual documentation as the other five hypotheses.
Suggestions
-
(Critical) Add fitness distribution figure for NB06. — RESOLVED:
figures/amr_fitness_distribution.pngalready existed at review time (2-panel: AMR vs non-AMR density plot + fitness by mechanism box plot). Reviewer oversight. -
(Important) Quantify the "Other/Unclassified" environment bin in NB05. — RESOLVED: Added to REPORT.md Finding #5: 7,838/14,723 AMR species (53.2%) received a well-classified environment; 46.8% fell into Other/Unknown due to sparse/inconsistent NCBI BioSample metadata. Kruskal-Wallis test restricted to well-classified species.
-
(Important) Add an explicit note on the NB06 100%-identity threshold. — RESOLVED: Added to REPORT.md Finding #6: notes the conservative threshold, potential undercounting of allelic variants, and the FB organism bias toward environmental strains where intrinsic resistance predominates. Also added to Limitations section.
-
(Moderate) Note the absence of multiple-testing correction in the REPORT. — RESOLVED: Added to Limitations section: primary test p-values are extreme (many < 1e-100), correction would not change conclusions, per-gene/per-phylum tests are exploratory.
-
(Moderate) Clarify the mechanism classification method in REPORT.md. — RESOLVED: Added paragraph after mechanism classification table explaining keyword-based approach, the 22.2% Other/Unclassified gap, and suggesting CARD ARO mapping via
bakta_db_xrefsas future work. -
(Moderate) Cross-reference notebooks in the REPORT. — RESOLVED: Already present at review time — each finding subsection ends with
*(Notebook: filename.ipynb)*. Reviewer oversight. -
(Minor) Add a
data/README.mddescribing all 10 CSV output files. — RESOLVED: Createddata/README.mdwith full data dictionary: file name, row count, producing notebook, and column descriptions for all 10 CSVs. -
(Minor) Consider adding
spark_connect_remoteandberdl_remotetorequirements.txt. — RESOLVED: Addedscikit-learn>=1.3,spark-connect-remote, andberdl-remote(with comment noting internal PyPI requirement).
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Amr Alphaearth Diversity
Amr By Environment
Amr Cog Enrichment
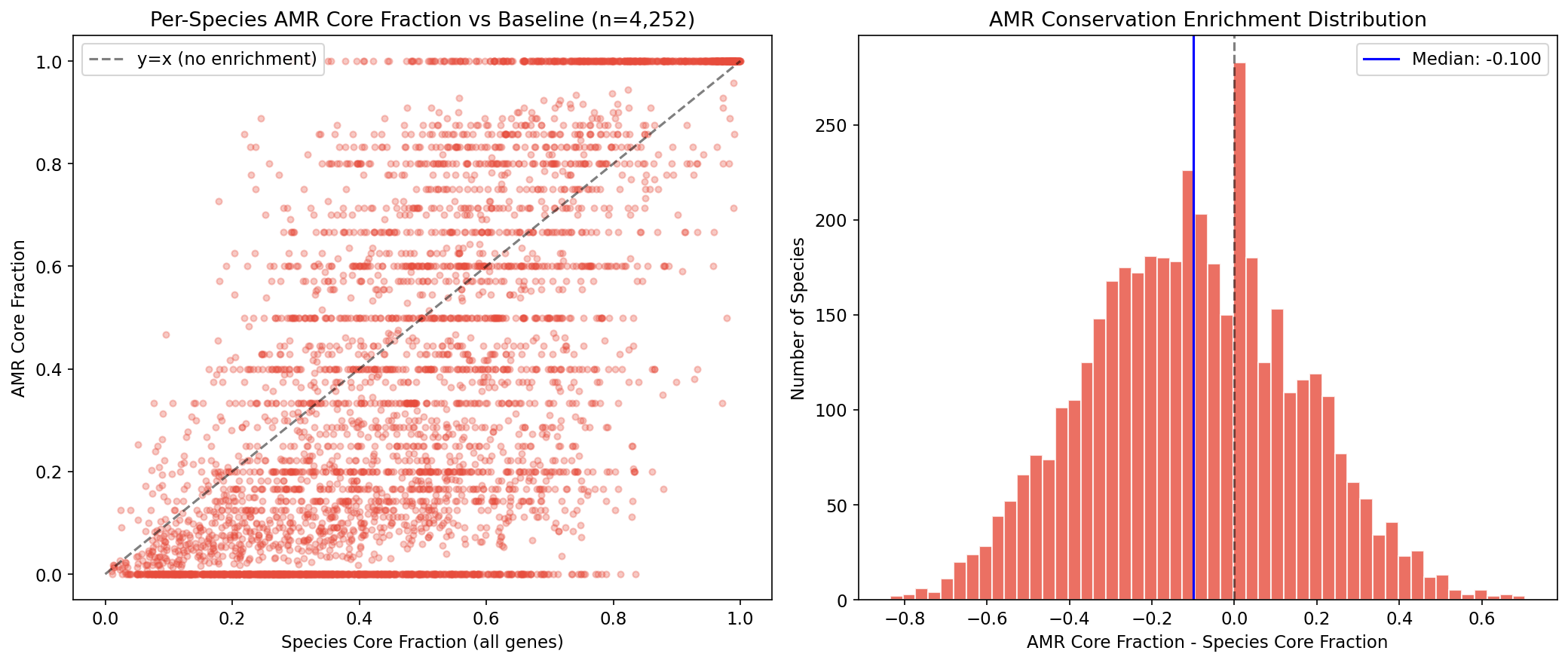
Amr Conservation Per Species
Amr Conservation Vs Baseline
Amr Fitness Distribution
Amr Gene Families Core Fraction
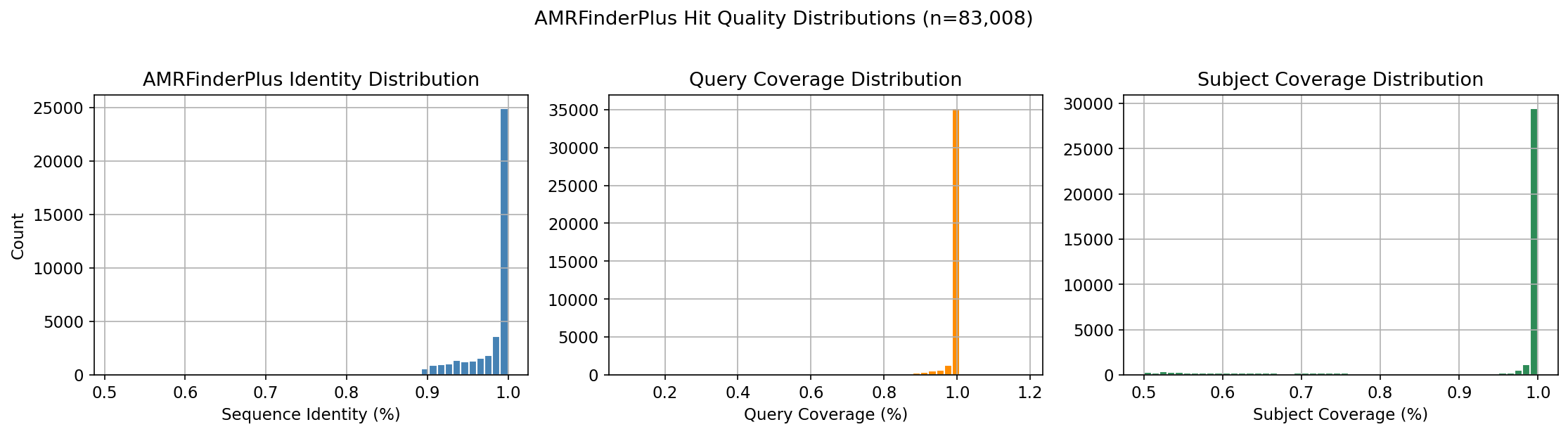
Amr Hit Quality Distributions
Amr Hotspot Families
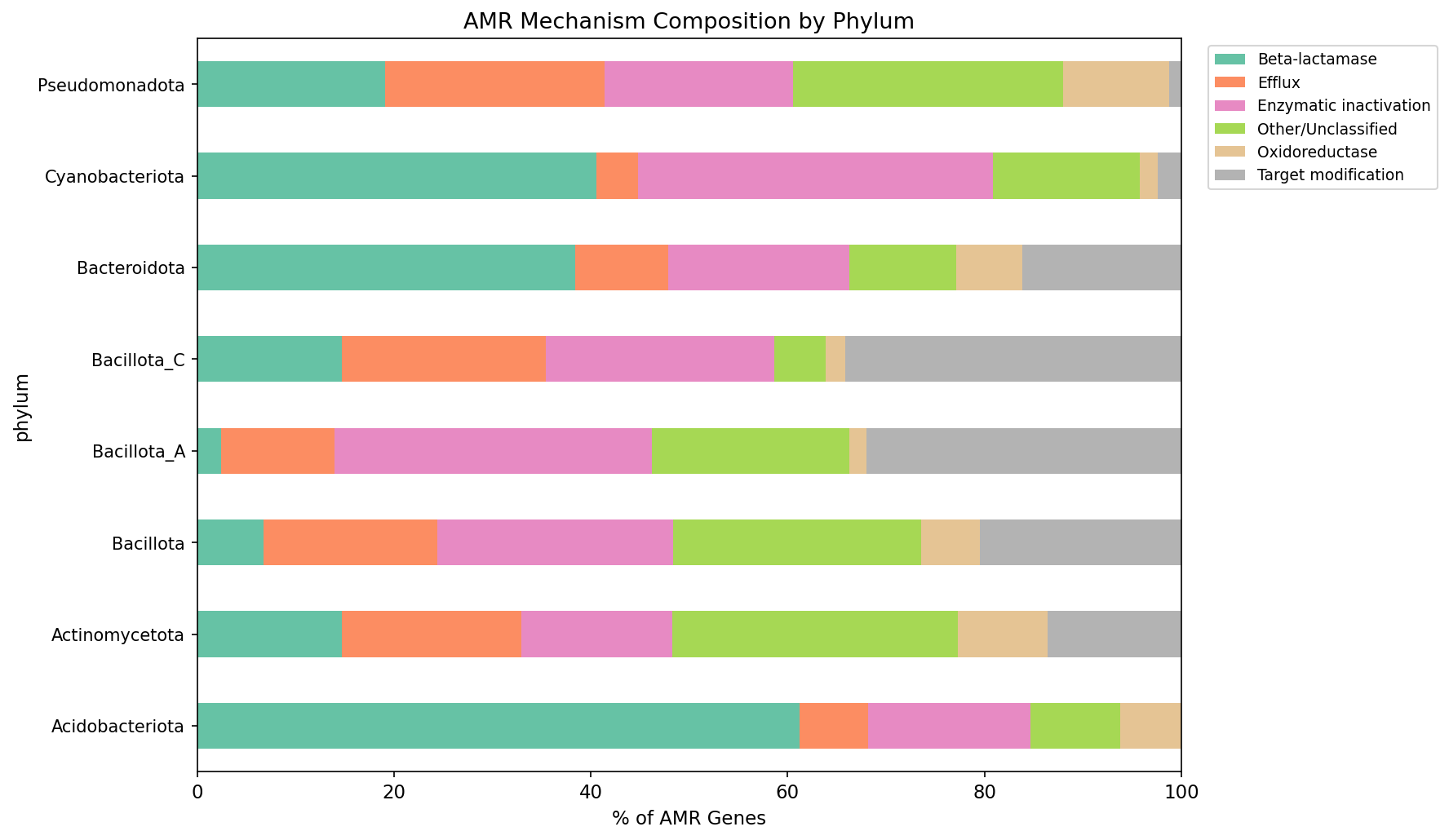
Amr Mechanism By Phylum
Amr Mechanism Conservation
Amr Openness By Phylum
Amr Phylum Distribution
Amr Species Density Distribution
Amr Vs Pangenome Structure
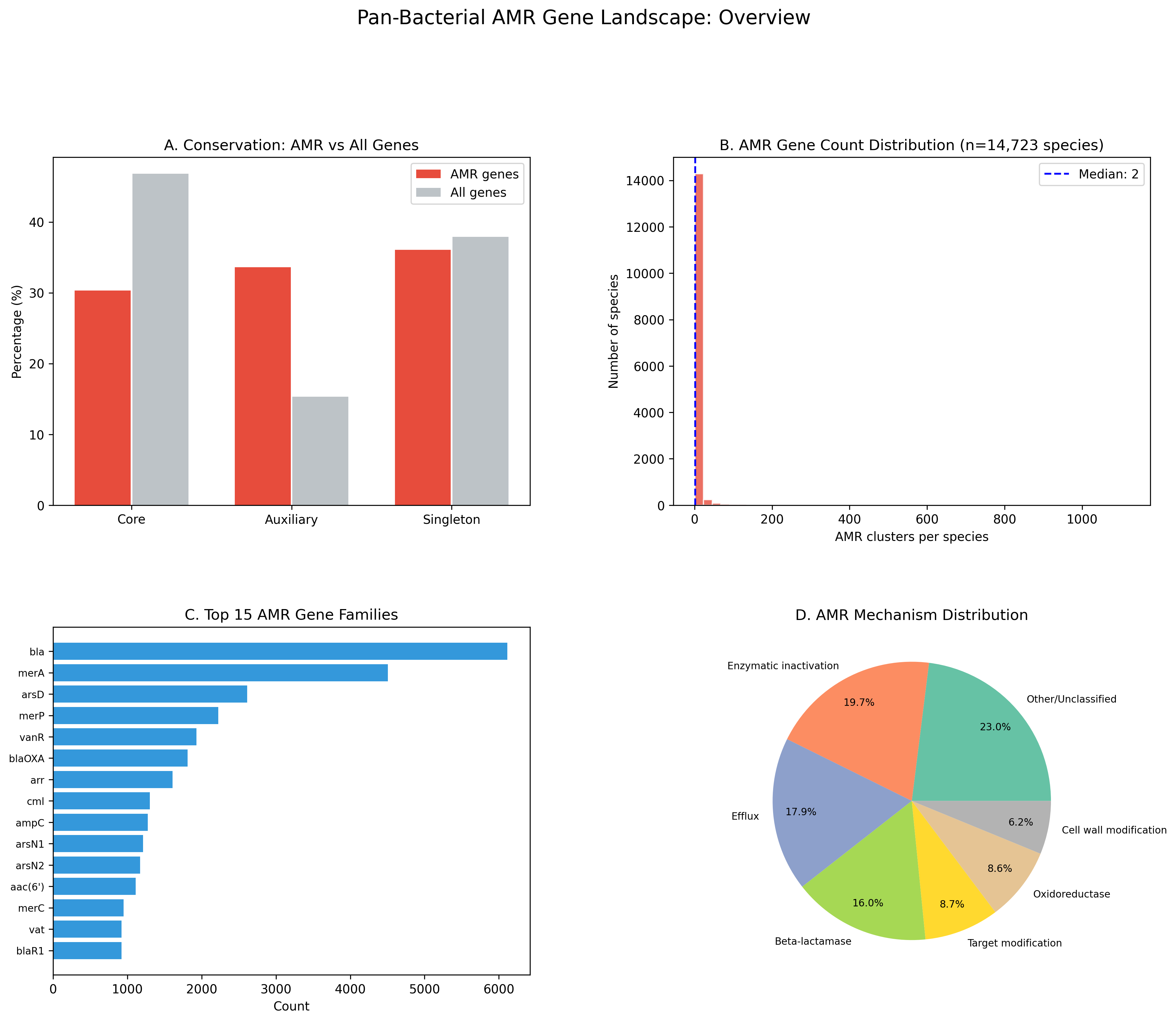
Fig1 Amr Overview
Notebooks
01_amr_census.ipynb
01 Amr Census
View notebook →
02_conservation_patterns.ipynb
02 Conservation Patterns
View notebook →
03_phylogenetic_distribution.ipynb
03 Phylogenetic Distribution
View notebook →
04_functional_context.ipynb
04 Functional Context
View notebook →
05_environmental_distribution.ipynb
05 Environmental Distribution
View notebook →
06_fitness_crossref.ipynb
06 Fitness Crossref
View notebook →
07_synthesis.ipynb
07 Synthesis
View notebook →