The Pan-Bacterial Essential Genome
CompletedResearch Question
Which essential genes are conserved across bacteria, which are context-dependent, and can we predict function for uncharacterized essential genes using module context from non-essential orthologs?
Research Plan
Hypothesis
- H0: Essential gene families are independent across organisms — essentiality in organism A does not predict essentiality of orthologs in organism B. Hypothetical essential genes cannot be functionally annotated via ortholog-module context.
- H1: A subset of essential gene families are universally essential across diverse bacteria, while others show context-dependent essentiality determined by genomic background. Non-essential orthologs in ICA fitness modules can transfer functional context to predict function for hypothetical essential genes.
Revision History
- v1 (2026-02-15): Initial plan — designed and executed in a single session. Covered all 48 FB organisms, 3 analysis notebooks, function prediction via module transfer.
Overview
This project clusters essential genes (defined by RB-TnSeq: no viable transposon mutants recovered) into cross-organism ortholog families and classifies them as universally essential, variably essential, or never essential. It builds on two upstream projects: conservation_vs_fitness (FB-pangenome link table) and fitness_modules (ICA modules and cross-organism families). For uncharacterized essential genes (~20% of all essentials are hypothetical), function is predicted by finding non-essential orthologs in other organisms that participate in ICA fitness modules, transferring the module's functional context. See RESEARCH_PLAN.md for the detailed hypothesis, query strategy, and analysis plan.
Key results: 15 families are essential in all 48 organisms (the irreducible core of bacterial life), variable essentiality is the norm (28% of families), and 7,084 orphan essential genes (58.7% hypothetical) represent the frontier of unknown biology. See REPORT.md for full findings and interpretation.
Key Findings
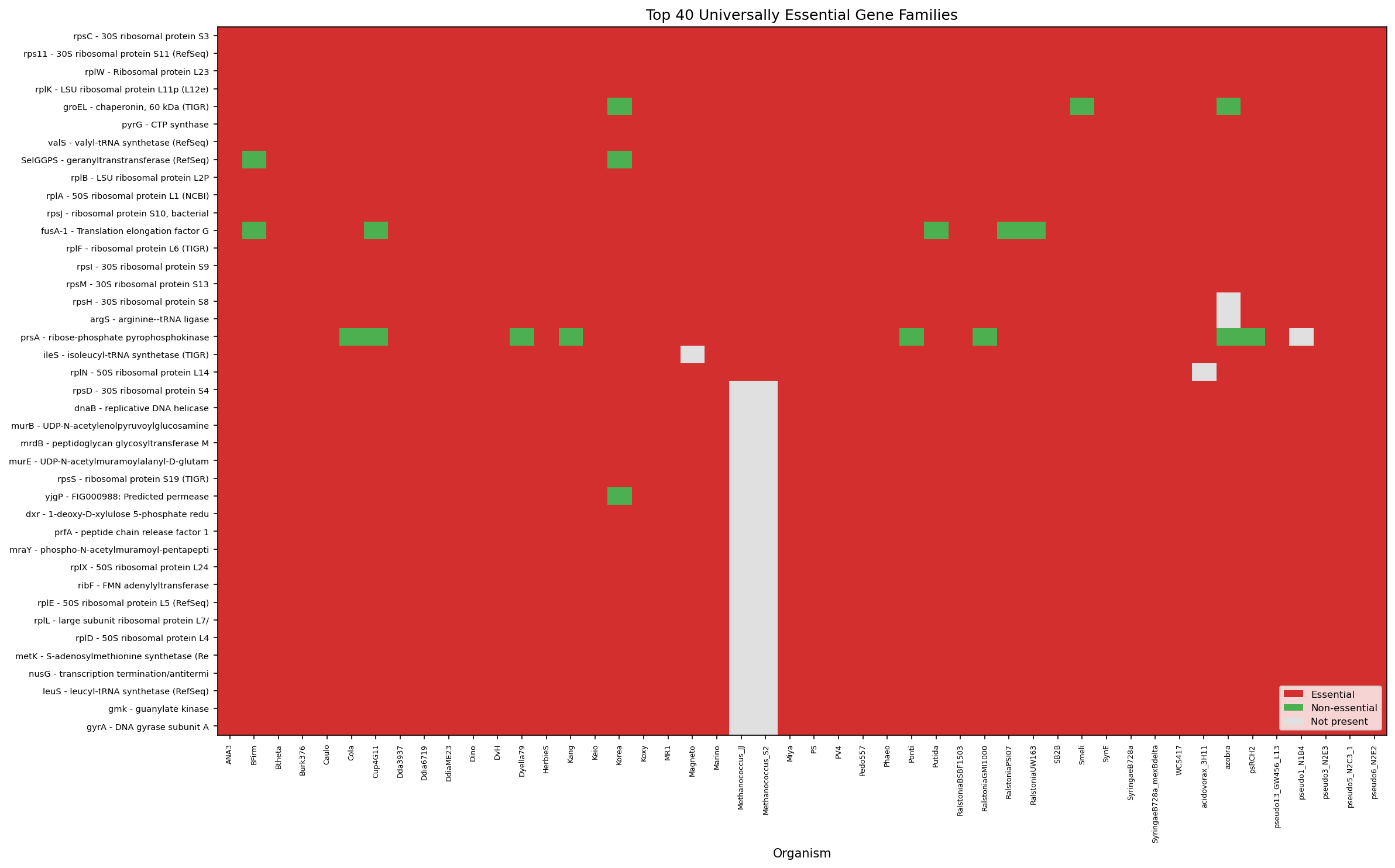
15 Gene Families Are Essential in All 48 Bacteria
The absolute core of bacterial life: ribosomal proteins (rpsC, rplW, rplK, rplB, rplA, rplF, rps11, rpsJ, rpsI, rpsM), chaperonin (groEL), CTP synthase (pyrG), translation elongation factor G (fusA), valyl-tRNA synthetase (valS), and geranyltranstransferase (SelGGPS). These 15 families span all 48 organisms with no exceptions.
(Notebook: 02_essential_families.ipynb)
Only 5% of Ortholog Families Are Universally Essential
Of 17,222 ortholog families across 48 bacteria, 859 (5.0%) are universally essential. Of these, 839 are strict single-copy families (copy ratio <=1.5, no non-essential paralogs); 20 contain paralogs. 4,799 families (27.9%) are variably essential -- essential in some organisms, non-essential in others. 11,564 (67.1%) are never essential.
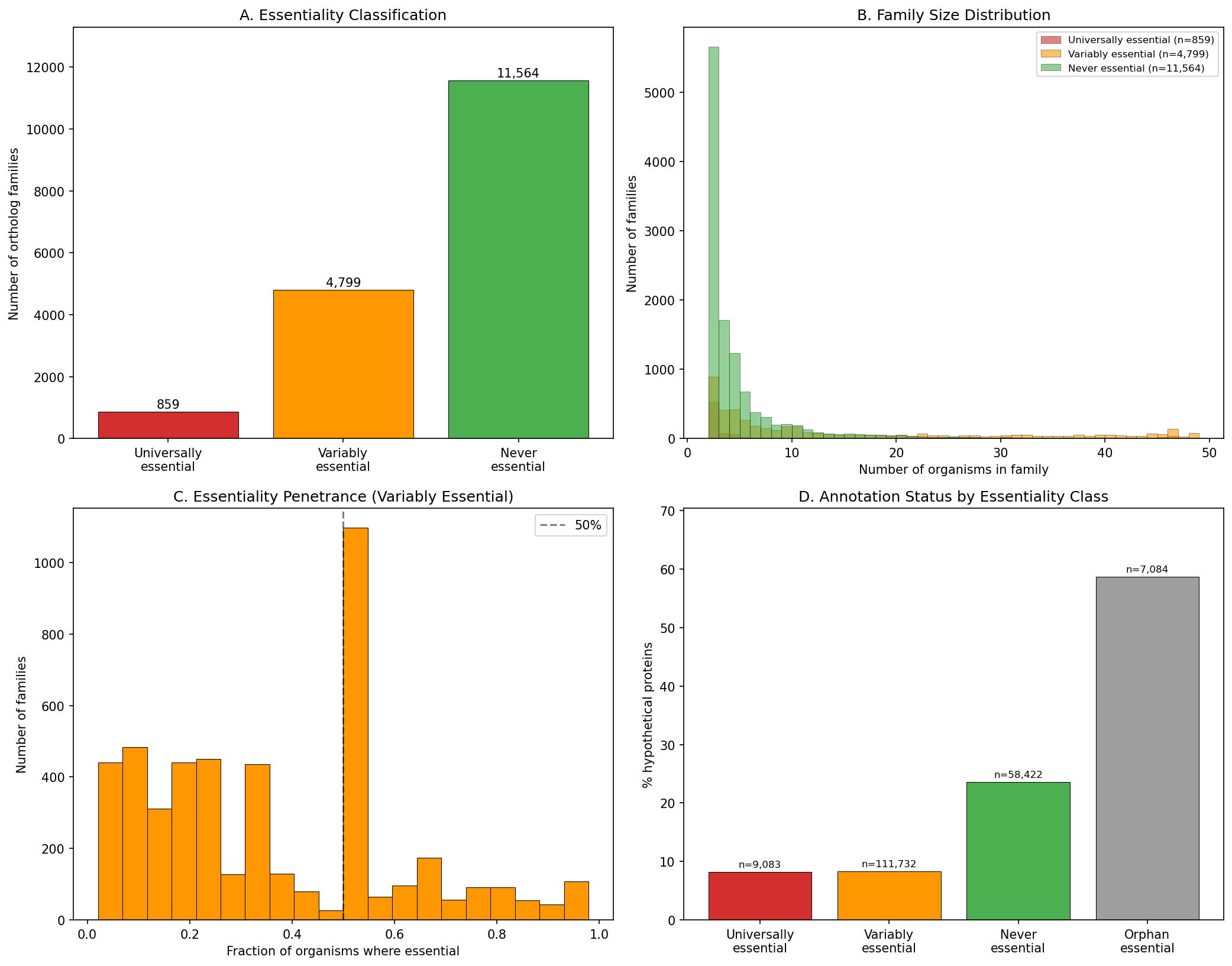
(Notebook: 02_essential_families.ipynb)
Orphan Essential Genes Are 58.7% Hypothetical
7,084 essential genes have no orthologs in any other FB organism. These are 58.7% hypothetical -- the least characterized yet most functionally important genes in each organism. By contrast, universally essential genes are only 8.2% hypothetical.
(Notebook: 02_essential_families.ipynb)
1,382 Function Predictions for Hypothetical Essentials
Module transfer via non-essential orthologs in ICA fitness modules generated 1,382 function predictions for hypothetical essential genes (35.3% of predictable targets). All predictions are family-backed. Top predicted functions: TIGR00254 (signal transduction), PF00460 (flagellar basal body rod), PF00356 (lactoylglutathione lyase).
(Notebook: 03_function_prediction.ipynb)
Universally Essential Families Are Overwhelmingly Core
Universally essential genes are 91.7% core vs 80.7% for non-essential. 71% of universally essential families are 100% core across all genomes in their species. Orphan essentials are only 49.5% core -- strain-specific essential functions.
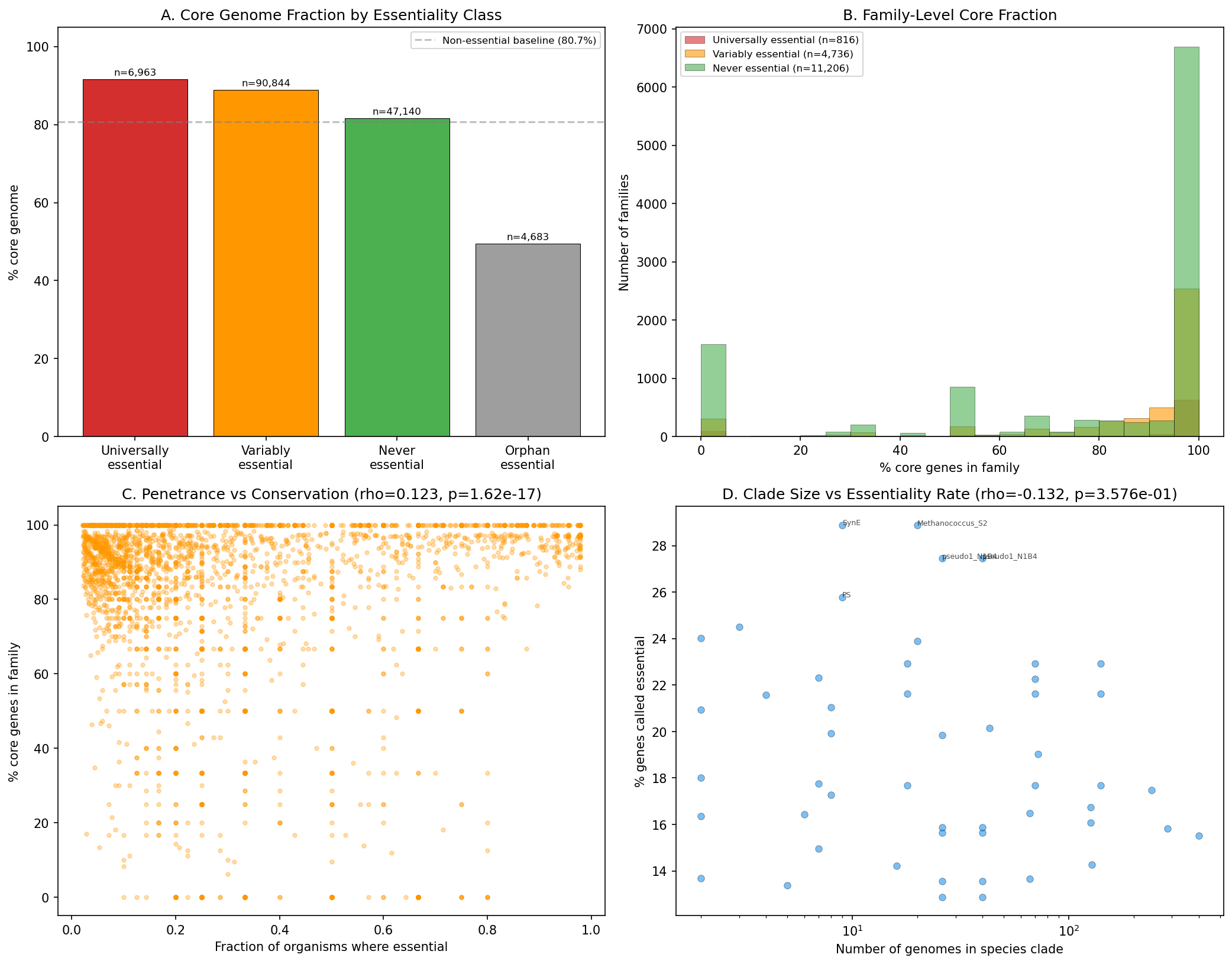
(Notebook: 04_conservation_architecture.ipynb)
Results
Essential Gene Landscape
- 221,005 genes across 48 organisms, 41,059 essential (18.6%)
- Essentiality rate ranges from 12.2% (Pedo557) to 29.7% (Magneto)
- 2,838,750 BBH pairs yield 17,222 ortholog groups spanning all 48 organisms
- Essential genes are shorter than non-essential (median 675 bp vs 885 bp); 17.8% of essentials are <300 bp
Family Classification
| Class | Families | % | Description |
|---|---|---|---|
| Universally essential | 859 | 5.0% | Essential in every organism where family has members |
| Variably essential | 4,799 | 27.9% | Essential in some organisms, not others |
| Never essential | 11,564 | 67.1% | No essential members in any organism |
Variably essential families have median essentiality penetrance of 33%. 813 families are >50% essential, 704 are <10% essential.
Conservation Hierarchy
| Essentiality class | % Core | n genes |
|---|---|---|
| Universally essential | 91.7% | 6,963 |
| Variably essential | 88.9% | 90,844 |
| Never essential | 81.7% | 47,140 |
| Orphan essential | 49.5% | 4,683 |
Weak positive correlation between essentiality penetrance and core fraction (rho=0.123, p=1.6e-17): families essential in more organisms tend to be slightly more core (97.1% for >80% penetrance vs 92.8% for <20%). Clade size does NOT predict essentiality rate (rho=-0.13, p=0.36).
Function Predictions
- 8,297 hypothetical essential genes across 48 organisms (20.2% of all essentials)
- 3,912 have orthologs (predictable); 4,385 are orphans (not predictable by this method)
- 1,382 predictions generated (35.3% of predictable targets), all family-backed
- Predictions span 48 organisms, from 90 predictions (pseudo1_N1B4) to 1 (Caulo)
Interpretation
The Essential Genome Is Small and Deeply Conserved
Only 859 of 17,222 ortholog families (5%) are universally essential, and just 15 are essential in all 48 organisms. These 15 families -- ribosomal proteins, groEL, CTP synthase, translation elongation factor G, and valyl-tRNA synthetase -- represent the irreducible functional core of bacterial life. This aligns with Koonin (2003), who estimated ~60 proteins are common to all cellular life, and Gil et al. (2004), who proposed a minimal gene set of ~206 genes. Our experimentally-defined universally essential set is smaller because it is restricted to genes where transposon disruption is lethal across all 48 tested organisms, which is more stringent than computational conservation.
Variable Essentiality Is the Norm, Not the Exception
The largest category is variably essential families (4,799, 28%) -- genes essential in some organisms but dispensable in others. With a median essentiality penetrance of 33%, most essential gene families are essential in only a minority of organisms. This directly extends Rosconi et al. (2022), who demonstrated within S. pneumoniae that the pan-genome makes gene essentiality "strain-dependent and evolvable." Our analysis shows this principle holds across 48 phylogenetically diverse species spanning Proteobacteria, Bacteroidetes, Firmicutes, and Archaea.
Variable essentiality implies that whether a gene is essential depends on genomic context -- the presence of paralogs, alternative pathways, or compensatory functions in the accessory genome. This has implications for antibiotic target selection: only the 859 universally essential families are reliable broad-spectrum targets.
Orphan Essentials: The Frontier of Unknown Biology
7,084 essential genes have no detectable orthologs in any other Fitness Browser organism. These orphan essentials are 58.7% hypothetical -- the highest uncharacterized fraction of any category. They represent strain-specific essential functions that may include recently acquired genes, rapidly evolving proteins, or lineage-specific innovations. Their low core genome rate (49.5%) suggests many are not conserved even within their own species. Hutchison et al. (2016) similarly found that 149 of 473 genes (31%) in their minimal Mycoplasma genome had unknown function -- our analysis shows this "dark matter" of essential genes is even larger when considering diverse bacteria.
Module Transfer Illuminates Essential Gene Function
The ortholog-module transfer approach generated 1,382 function predictions for hypothetical essential genes. The method works because essential genes are invisible to ICA (no fitness data), but their non-essential orthologs in other organisms DO have fitness data and can participate in modules. By transferring the module's functional context across organisms, we bridge the gap between essentiality (which prevents fitness measurement) and function prediction (which requires fitness data).
Literature Context
- Koonin (2003) estimated ~60 universally conserved proteins across all life, primarily translation-related. Our 15 pan-bacterial essential families are consistent -- dominated by ribosomal proteins and translation machinery, but also include cell wall biosynthesis and nucleotide metabolism genes that may be bacteria-specific essentials.
- Gil et al. (2004) proposed a ~206-gene minimal bacterial gene set based on computational comparison of reduced genomes. Our 859 universally essential families is larger because it includes genes essential in free-living bacteria with larger genomes, not just obligate symbionts with minimal genomes.
- Rosconi et al. (2022) demonstrated context-dependent essentiality across 36 S. pneumoniae strains. Our work extends this concept from within-species to cross-species variation: the same gene can be essential in Methanococcus but dispensable in Pseudomonas, depending on metabolic context.
- Duffield et al. (2010) identified 52 conserved essential proteins across 14 bacterial genomes as drug targets. Our approach is complementary but uses experimental (RB-TnSeq) essentiality data across 48 organisms rather than computational prediction, providing empirical validation of conservation patterns.
- Price et al. (2018) generated the Fitness Browser data used here. Our work adds a cross-organism essentiality dimension to their fitness data, enabling function prediction for essential genes that are outside the reach of fitness profiling.
- Hutchison et al. (2016) found 149 essential genes of unknown function in their synthetic minimal genome. Our 7,084 orphan essentials (58.7% hypothetical) show this problem is universal across bacteria, not specific to Mycoplasma.
Limitations
- Essential gene definition is an upper bound: Genes without transposon insertions may lack insertions due to small size, AT-rich sequence, or scaffold edge effects, not true essentiality.
- BBH orthology is conservative: Bidirectional best BLAST hits miss paralogs, gene fusions, and distant homologs. Some "orphan essentials" may have undetected orthologs with diverged sequences.
- Connected components can over-merge: Transitive connections in the BBH graph can merge unrelated genes into the same ortholog group, particularly for multi-domain proteins.
- Essentiality is condition-dependent: RB-TnSeq essentiality is defined under specific library construction conditions (typically rich media). Genes essential only under stress would be missed.
- 48 organisms is taxonomically limited: The Fitness Browser organisms are biased toward culturable Proteobacteria. Essentiality patterns in uncultured lineages, Actinobacteria, or Firmicutes are underrepresented.
- Module transfer predictions are indirect: Function predictions for essential genes come from non-essential orthologs in other organisms. The essential gene's function may have diverged.
Future Directions
- Characterize the 7,084 orphan essentials -- Are they on mobile elements? Recently acquired? Rapidly evolving? What functional categories do they represent?
- Validate predictions experimentally -- The 1,382 module-transfer predictions could be tested via CRISPRi knockdown under specific conditions predicted by the module context.
- Connect to metabolic networks -- For variably essential families, determine whether essentiality correlates with metabolic pathway completeness (e.g., is a gene essential only when an alternative pathway is absent?).
- Expand taxonomic coverage -- As the Fitness Browser adds more organisms, re-run the analysis to test whether the 15 pan-essential families remain universal.
- Compare to DEG database -- Cross-reference with the Database of Essential Genes to identify families that are essential in non-FB organisms.
Data
Sources
| Dataset | Description | Source |
|---|---|---|
| Fitness Browser gene table | 221,005 genes across 48 organisms | Price et al. (2018) |
| FB BBH pairs | Bidirectional best BLAST hits | Fitness Browser besthit table |
| KBase pangenome link table | Gene-to-cluster conservation mapping | conservation_vs_fitness/data/fb_pangenome_link.tsv |
| ICA fitness modules | Co-regulated gene modules | fitness_modules/data/modules/ |
| Module families | Cross-organism module families | fitness_modules/data/module_families/ |
| SEED annotations | Functional category assignments | conservation_vs_fitness/data/seed_annotations.tsv |
Generated Data
| File | Description |
|---|---|
data/all_essential_genes.tsv |
Essential status for 221,005 genes across 48 organisms |
data/all_bbh_pairs.csv |
2.84M BBH pairs across 48 organisms |
data/all_ortholog_groups.csv |
179,237 gene-OG assignments across 17,222 ortholog groups |
data/essential_families.tsv |
Per-family essentiality classification (17,222 families) |
data/essential_predictions.tsv |
1,382 function predictions for hypothetical essential genes |
data/family_conservation.tsv |
Per-family pangenome conservation metrics (16,758 families with links) |
References
- Price MN et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557:503-509. DOI: 10.1038/s41586-018-0124-0. PMID: 29769716
- Parks DH et al. (2022). "GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy." Nucleic Acids Res 50:D199-D207. PMID: 34520557
- Rosconi F et al. (2022). "A bacterial pan-genome makes gene essentiality strain-dependent and evolvable." Nat Microbiol 7:1580-1592. DOI: 10.1038/s41564-022-01208-7. PMID: 36097170
- Hutchison CA 3rd et al. (2016). "Design and synthesis of a minimal bacterial genome." Science 351:aad6253. DOI: 10.1126/science.aad6253. PMID: 27013737
- Koonin EV. (2003). "Comparative genomics, minimal gene-sets and the last universal common ancestor." Nat Rev Microbiol 1:127-136. PMID: 15035042
- Gil R et al. (2004). "Determination of the core of a minimal bacterial gene set." Microbiol Mol Biol Rev 68:518-537. PMID: 15353568
- Duffield M et al. (2010). "Predicting conserved essential genes in bacteria." Mol BioSyst 6:2482-2489. PMID: 20949199
Discoveries
15 gene families are essential in ALL 48 bacteria
February 2026Cross-organism essential gene analysis across 48 diverse bacteria (221,005 genes, 17,222 ortholog families) reveals 15 families that are essential in every organism: ribosomal proteins (rpsC, rplW, rplK, rplB, rplA, rplF, rps11, rpsJ, rpsI, rpsM), chaperonin (groEL), CTP synthase (pyrG), translation
Read more →Only 5% of ortholog families are universally essential
February 2026Of 17,222 ortholog families across 48 bacteria, only 859 (5.0%) are universally essential — essential in every organism where the family has members. 4,799 families (27.9%) are variably essential (essential in some organisms, non-essential in others), and 11,564 (67.1%) are never essential. Variable
Read more →7,084 essential genes have no orthologs in any other FB organism. Of these, 58.7% are annotated as hypothetical proteins — the least characterized yet most functionally important genes in each organism. By contrast, universally essential genes are only 8.2% hypothetical. The orphan essentials are pr
Read more →Universally essential genes are 91.7% core vs 80.7% for non-essential genes. 71% of universally essential families are 100% core across all genomes in their species. Orphan essentials (no orthologs) are only 49.5% core — they are strain-specific essential functions that haven't been conserved across
Read more →By finding non-essential orthologs that participate in ICA fitness modules, we generated 1,382 function predictions for hypothetical essential genes across 48 organisms. All predictions are backed by cross-organism module family conservation. This demonstrates that module context from non-essential
Read more →Data Collections
Derived Data
This project builds on processed data from other projects.
Used By
Data from this project is used by other projects.
Review
Summary
This is an exceptionally well-executed project that classifies essential gene families across 48 Fitness Browser organisms and predicts function for uncharacterized essential genes via ICA module transfer. The project builds logically on two upstream projects (conservation_vs_fitness, fitness_modules) and synthesizes their data into a novel analysis with biologically meaningful results: 859 universally essential families (15 spanning all 48 organisms), 4,799 variably essential families, 1,382 function predictions for hypothetical essentials, and a clear conservation hierarchy linking essentiality breadth to pangenome core status. The README is outstanding — thorough methodology, well-contextualized findings with six cited references, honest limitations, and actionable future directions. All three notebooks have saved outputs in every code cell, three publication-quality multi-panel figures are generated, and all intermediate data files are cached. The main areas for improvement are minor: NB03 (function prediction) lacks any visualizations, the confidence scoring formula is undocumented, and the 859→816 universally essential family count difference between NB02 and NB04 is unexplained in the notebooks.
Methodology
Research question: Clearly stated, multi-part, and testable — classifying essential gene families along a universality spectrum and predicting function for hypothetical essentials via ortholog-module transfer. The question builds logically on two predecessor projects and fills a genuine gap.
Approach: The five-step pipeline (Spark extraction → ortholog groups → classify families → predict function → conservation architecture) is well-structured. The Spark-dependent extraction is cleanly separated into src/extract_data.py, while all three downstream notebooks run locally from cached data — a good architectural pattern that follows the pitfalls.md recommendation for checkpointing.
Data sources: Clearly documented in a table in the README with specific database tables and their uses. Cross-project dependencies are explicitly listed (9 files across 2 upstream projects) with file paths and source project names.
Essential gene definition: Correctly defined as CDS genes (type=1) absent from genefitness, matching the method documented in pitfalls.md ("FB Gene Table Has No Essentiality Flag"). The README appropriately acknowledges this is an upper bound on true essentiality. The gene length analysis in NB02 shows 17.8% of essential genes are <300 bp, which is flagged as a potential false positive source but no filter is applied — a reasonable choice with the caveat documented in Limitations.
Ortholog grouping: Connected components of the BBH graph is standard. The README acknowledges the over-merging limitation. The code computes copy_ratio and is_single_copy flags (NB02), and the README carefully distinguishes 839 strict single-copy from 20 multi-copy universally essential families.
Reproducibility: Strong. The README includes a ## Reproduction section with exact commands. requirements.txt lists all six dependencies with minimum versions. Spark vs local steps are clearly distinguished. The data/.gitignore correctly excludes large regenerable files (all_bbh_pairs.csv at 144M, all_essential_genes.tsv at 22M, all_seed_annotations.tsv at 12M) while keeping derived analysis outputs in version control.
Code Quality
SQL queries (extract_data.py): Correct and well-structured. Uses CAST(begin AS INT) and CAST(end AS INT) consistent with the pitfall that all FB columns are strings. The type = '1' filter correctly uses string comparison. Queries properly filter by orgId for the large genefitness table. The BBH extraction filters both orgId1 and orgId2 to the 48 target organisms, correctly addressing the "Ortholog Scope Must Match Analysis Scope" pitfall.
Notebook organization: All three notebooks follow the pattern: markdown header → imports/data loading → computation → output/visualization. Markdown cells clearly delineate sections. Data is loaded from cached files, not re-queried.
Notebook outputs: All 24 code cells across 3 notebooks have saved outputs (10/10, 7/7, 7/7). This is excellent — readers can inspect all intermediate results without re-running.
Boolean handling: Defensive and correct. Every notebook converts is_essential via astype(str).str.strip().str.lower() == 'true', handling TSV round-tripping safely. The in_og flag in NB02 uses .fillna(False).astype(bool), correctly addressing the documented pitfall about fillna(False) producing object dtype. The assertion assert essential['in_og'].sum() > 30000 guards against silent merge failure.
Merge-based approach: NB02 and NB03 use merge-based operations for key lookups (e.g., essential.merge(og_keys, ...) in NB02), avoiding the slow row-wise .apply() pattern documented in pitfalls.md.
Pitfall awareness: The project correctly handles: (1) essential genes being invisible in genefitness (foundation of the analysis); (2) string-typed columns with CAST; (3) ortholog scope matching analysis scope; (4) fillna(False).astype(bool) for boolean columns; (5) seedannotation for functional descriptions (not seedclass); (6) file-based caching for Spark queries; (7) berdl_notebook_utils.setup_spark_session import for CLI Spark access.
Potential issues:
-
Graph construction via iterrows() (extract_data.py, lines 128-131): Building the NetworkX graph iterates over 2.84M BBH pairs with
iterrows(). This works but is slow; vectorized construction (e.g.,G.add_edges_from(zip(node1_series, node2_series))) would be 10-100x faster. Since results are cached to disk, this is acceptable but worth noting for re-runs. -
Family classification loop (NB02, cell
842be4b5): The per-familyforloop over 17,222 OGs is functional but could benefit from vectorized groupby aggregation. Again, results are cached, so this is minor. -
NB04 family count discrepancy: NB02 classifies 859 universally essential families, but NB04's conservation analysis includes only 816 (579 are 100% core, 670 are ≥90% core). The difference (43 families) presumably arises because only 44 of 48 organisms have pangenome links in the
fb_pangenome_link.tsvtable. This gap is not documented in NB04, which could confuse readers comparing numbers across notebooks.
Findings Assessment
Well-supported conclusions:
- The 15 pan-bacterial essential families (ribosomal proteins, groEL, fusA, pyrG, valS) are biologically plausible and consistent with Koonin (2003). The heatmap confirms these span all 48 organisms with red (essential) across every column.
- The conservation hierarchy (universally essential 91.7% > variably essential 88.9% > never essential 81.7% > orphan essential 49.5% core) is clearly demonstrated in NB04 outputs and Figure 3.
- Variable essentiality is quantified well: median penetrance of 33%, 813 families >50% essential, 704 families <10% essential.
- The 839 single-copy vs 20 multi-copy distinction among universally essential families effectively addresses over-merging concerns.
- 1,382 function predictions via module transfer — all family-backed — represent a novel and methodologically sound contribution.
- The observation that orphan essentials are 58.7% hypothetical vs 8.2% for universally essential genes is striking and well-supported.
Literature integration: Excellent. Six papers are cited in the Interpretation section with specific quantitative comparisons (Koonin's ~60 universal proteins vs 15 pan-essential families, Gil et al.'s ~206-gene minimal set vs 859 families, Rosconi et al.'s within-species results extended to cross-species). The references.md has 9 proper citations with DOIs and PMIDs.
Limitations: Unusually thorough — six specific limitations covering false positive essentials, BBH conservatism, connected-component over-merging, condition-dependent essentiality, taxonomic bias, and indirect function predictions.
Visualizations: Three well-designed multi-panel figures. The overview figure (4-panel: family counts, size distribution, penetrance histogram, annotation status) effectively summarizes the classification. The heatmap clearly shows essentiality patterns across 48 organisms with a consistent color scheme. The conservation architecture figure (4-panel) includes the penetrance-vs-conservation scatter and the clade-size analysis. All figures have proper axis labels, legends, and sample size annotations.
Gap: NB03 (function prediction) generates no figures despite producing 1,382 predictions. This is the weakest notebook for visual communication.
Suggestions
-
Add visualizations to NB03 (medium priority): The function prediction notebook produces 1,382 predictions but has no figures. Consider adding: (a) a histogram of prediction confidence scores, (b) a bar chart of the top 10-15 most common predicted functional terms, or (c) a scatter/heatmap showing which organisms provide predictions vs which receive them. This would make the notebook self-contained for visual inspection.
-
Document the 859→816 family count gap in NB04 (low priority): Add a brief note in NB04 explaining that the universally essential family count drops from 859 to 816 because 4 of 48 organisms lack pangenome link coverage. A single print statement (e.g.,
print(f"Families with conservation data: {len(fam_cons[fam_cons['essentiality_class']=='universally_essential'])} / {len(families[families['essentiality_class']=='universally_essential'])}")) would suffice. -
Document the confidence scoring formula (low priority): The NB03 confidence score (
-log10(FDR) + log10(family_breadth)) is computed in code but not explained in any markdown cell. Adding a brief description of the formula and its rationale would help readers interpretessential_predictions.tsv. -
Consider a gene length sensitivity analysis (low priority, future work): The analysis flags 17.8% of essentials as <300 bp (potential false positives). A supplementary cell in NB02 showing how family classification changes with a 300 bp minimum filter would quantify the impact and strengthen the core findings.
-
Optimize graph construction for re-runs (nice-to-have): Replace the
iterrows()loop inextract_data.py(lines 128-131) with vectorized edge construction for the 2.84M BBH pairs. Since results are cached, this only matters for fresh extractions, but it would reduce the extraction step from minutes to seconds. -
Add a summary statistics cell to NB04 (nice-to-have): NB04 loads the link table and reports "177,863 gene-cluster links, 44 organisms" — noting that only 44 of 48 organisms have links. A follow-up cell listing which 4 organisms lack pangenome coverage would document the scope limitation upfront.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Conservation Architecture
Essential Families Heatmap
Essential Families Overview
Notebooks
Data Files
| Filename | Size |
|---|---|
all_ortholog_groups.csv |
5132.7 KB |
essential_families.tsv |
2800.3 KB |
essential_predictions.tsv |
212.2 KB |
family_conservation.tsv |
1328.8 KB |