The Pan-Bacterial Essential Metabolome
CompletedResearch Question
Which biochemical reactions are universally essential across bacteria, and what does the essential metabolome reveal about the minimal core metabolism required for microbial life?
Research Plan
Hypothesis
- H0: There is no conserved set of universally essential biochemical reactions; essential metabolism is highly species-specific.
- H1: A core set of biochemical reactions is universally essential across bacteria, concentrated in central carbon metabolism, amino acid biosynthesis, nucleotide metabolism, and core biosynthetic pathways.
Approach
Data Sources
| Source | Purpose | Scale | Filter Strategy |
|---|---|---|---|
projects/essential_genome/data/essential_families.tsv |
Universally essential gene families (859 families) | 859 families, 48 organisms | Precomputed, reuse via MinIO |
kbase_ke_pangenome.eggnog_mapper_annotations |
EC numbers for gene clusters | 93M annotations | Join on gene_cluster_id from essential families |
kbase_msd_biochemistry.reaction |
Biochemical reactions | 56K reactions | Filter by EC numbers from essential genes |
kbase_msd_biochemistry.reaction_ec |
EC → reaction mappings | Small | Join table for EC lookups |
Workflow
This project tests the new local BERDL workflow with both MinIO data access and Spark Connect queries.
MinIO Operations (test data download/upload):
1. Download existing essential gene data: mc cp --recursive berdl-minio/.../microbialdiscoveryforge/projects/essential_genome/data/ ./data/
2. Upload results: python tools/lakehouse_upload.py essential_metabolome
Spark Connect Queries (test local query execution):
1. Extract EC annotations for essential gene clusters
2. Join EC numbers to ModelSEED reactions
3. Query reaction properties (reversibility, pathways, cofactors)
Analysis Plan
Notebook 1: Data Extraction & Linking (01_data_extraction.ipynb)
Goal: Map essential gene families → EC numbers → biochemical reactions
Steps:
1. Load essential families from MinIO or local cache (essential_genome/data/essential_families.tsv)
2. Extract gene cluster IDs for universal essential families (859 families present in all 48 organisms)
3. Query eggnog_mapper_annotations to get EC numbers for essential clusters (Spark Connect)
4. Query kbase_msd_biochemistry to map EC → reactions (Spark Connect)
5. Filter to reactions with complete GPR associations (all constituent genes are essential)
Expected output:
- data/essential_gene_clusters.tsv — gene clusters in universal essential families
- data/essential_ec_numbers.tsv — EC numbers from essential genes
- data/essential_reactions.tsv — reactions catalyzed by essential genes
- data/universal_essential_reactions.tsv — reactions present in all 48 organisms
Spark Connect test: Medium-scale join (859 families → ~5K gene clusters → ~2K EC numbers → ~1K reactions)
Notebook 2: Metabolic Network Analysis (02_metabolic_analysis.ipynb)
Goal: Characterize the essential metabolome and test hypotheses
Analyses:
- Reaction coverage:
- How many reactions are universally essential? (all 48 organisms)
- How many are essential in ≥90% of organisms? ≥50%?
-
Species-specific essential reactions
-
Pathway enrichment:
- Which ModelSEED pathways are enriched in essential reactions?
- Compare to background (all annotated reactions)
-
Fisher's exact test with FDR correction
-
Cofactor dependency:
- Frequency of ATP, NAD+, NADP+, CoA, FAD in essential vs non-essential reactions
-
Chi-squared test for enrichment
-
Reaction properties:
- Reversibility (reversible vs irreversible)
- Stoichiometry complexity (number of reactants/products)
-
Reaction class distribution (oxidoreductase, transferase, hydrolase, etc.)
-
Network analysis:
- Build metabolic network from ModelSEED (nodes = compounds, edges = reactions)
- Calculate betweenness centrality for essential vs non-essential reactions
- Identify critical connectors in the metabolic network
Expected outputs:
- figures/reaction_coverage.png — histogram of reaction prevalence
- figures/pathway_enrichment.png — bar chart of enriched pathways
- figures/cofactor_dependency.png — cofactor frequency in essential reactions
- figures/network_centrality.png — centrality distributions
- data/pathway_enrichment.tsv — statistical test results
- data/reaction_properties.tsv — properties of essential vs non-essential reactions
Local analysis test: Network analysis (igraph/networkx), statistical tests (scipy), plotting (matplotlib/seaborn)
Performance Considerations
Spark queries:
- Essential gene cluster query: ~859 families → ~5K clusters (small, fast)
- eggNOG annotation join: ~5K clusters → ~93M table (indexed on query_name, should be fast)
- Biochemistry queries: Small tables (<100K rows), can scan or use exact lookups
Local analysis:
- Network analysis on ~1K essential reactions + ~10K background reactions (manageable)
- All datasets small enough for pandas/local processing after Spark extraction
Estimated runtime:
- NB01 (Spark extraction): 5-10 minutes
- NB02 (local analysis): 5-10 minutes
Revision History
- v1 (2026-02-17): Initial plan. Approach: link essential gene families from
essential_genomeproject to ModelSEED reactions via eggNOG EC annotations. Test local BERDL workflow (MinIO + Spark Connect). Two notebooks: extraction + analysis.
Overview
This pilot study analyzes metabolic pathway completeness across 7 bacterial organisms with essential gene data using GapMind predictions from kbase_ke_pangenome. The analysis reveals high conservation of amino acid biosynthesis pathways (17/18 pathways present in all organisms) with one notable exception: Desulfovibrio vulgaris lacks serine biosynthesis, representing a potential metabolic auxotrophy.
Key finding: Near-universal amino acid prototrophy among free-living bacteria, with organism-specific gaps reflecting ecological adaptation.
Limitation: E. coli genomes are absent from the pangenome GapMind dataset (too many genomes for species-level analysis), limiting this to a 7-organism pilot study rather than the intended pan-bacterial survey.
Key Findings
High Conservation of Amino Acid Biosynthesis Pathways
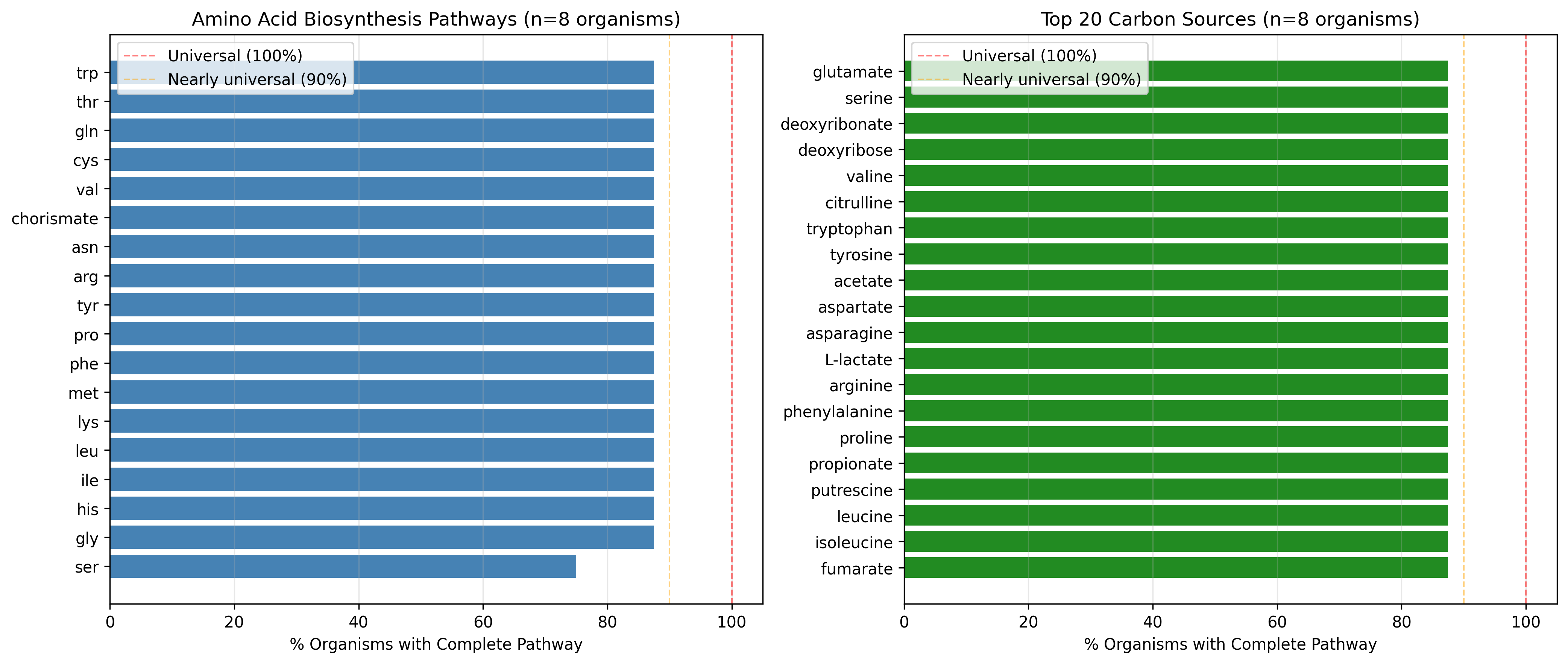
17 of 18 amino acid biosynthesis pathways are present in all 7 organisms analyzed (100% within this sample):
- Complete pathways: arg, asn, chorismate, cys, gln, gly, his, ile, leu, lys, met, phe, pro, thr, trp, tyr, val
One exception: Serine biosynthesis present in only 6 of 7 organisms (85.7%)
The missing organism is Desulfovibrio vulgaris, an anaerobic sulfate-reducing bacterium that appears to lack serine biosynthesis according to GapMind predictions.
(Notebook: 02_gapmind_pathway_analysis.ipynb)
Desulfovibrio vulgaris Serine Auxotrophy
Desulfovibrio vulgaris (DvH) is the only organism among the 7 that lacks a complete serine biosynthesis pathway according to GapMind (score_category: complete or likely_complete).
Biological context:
- D. vulgaris lives in anaerobic, organic-rich environments (sediments, intestinal tracts)
- Serine would be abundant from environmental protein degradation
- This could represent a genuine metabolic auxotrophy where the organism depends on external serine
Organism pathway completeness summary:
| Organism | Species | AA Pathways | % Complete |
|----------|---------|-------------|-----------|
| Caulo | Caulobacter vibrioides | 18/18 | 100% |
| MR1 | Shewanella oneidensis | 18/18 | 100% |
| PS | Pseudomonas aeruginosa | 18/18 | 100% |
| Putida | Pseudomonas putida | 18/18 | 100% |
| Smeli | Sinorhizobium meliloti | 18/18 | 100% |
| azobra | Azospirillum brasilense | 18/18 | 100% |
| DvH | Desulfovibrio vulgaris | 17/18 | 94.4% |
(Notebook: 02_gapmind_pathway_analysis.ipynb)
Conserved Carbon Source Utilization
Most widely conserved carbon sources (present in all 7 organisms, 87.5%):
- TCA cycle intermediates: fumarate, succinate
- Fermentation products: acetate, propionate, L-lactate
- Amino acids as carbon sources: All amino acids can be catabolized
- Nucleotide derivatives: deoxyribose, deoxyribonate
- Polyamines: putrescine
Nearly universal (6 of 7 organisms, 75%):
- ethanol, deoxyinosine
This pattern suggests a shared central catabolic capacity for breaking down common organic compounds, consistent with the diverse ecological niches these bacteria occupy.
(Notebook: 02_gapmind_pathway_analysis.ipynb)
GapMind Coverage Limitation Discovered
E. coli genomes are completely absent from GapMind in the KBase pangenome collection.
Coverage summary:
| Organism | Species | Genome ID | GapMind Predictions | Status |
|----------|---------|-----------|---------------------|--------|
| Keio | Escherichia coli K-12 | GCF_000005845.2 | 0 | ❌ Missing |
| DvH | Desulfovibrio vulgaris | GCF_000195755.1 | 694 | ✅ Present |
| MR1 | Shewanella oneidensis | GCF_000146165.2 | 694 | ✅ Present |
| Putida | Pseudomonas putida | GCF_000007565.2 | 1,041 | ✅ Present |
| PS | Pseudomonas aeruginosa | GCF_000006765.1 | 745 | ✅ Present |
| Caulo | Caulobacter vibrioides | GCF_000022005.1 | 694 | ✅ Present |
| Smeli | Sinorhizobium meliloti | GCF_000006965.1 | 1,735 | ✅ Present |
| azobra | Azospirillum brasilense | GCF_000011365.1 | 1,786 | ✅ Present |
Reason: E. coli was excluded from GTDB pangenome construction due to having too many genomes for species-level analysis.
Impact: This limits the current analysis to a pilot study with 7 organisms rather than the intended 45 FB organisms with essential gene data.
(Discovery: manual GapMind coverage checking)
Interpretation
Near-Universal Amino Acid Biosynthesis
The finding that 6 of 7 organisms (85.7%) possess complete biosynthesis pathways for all 18 amino acids suggests that amino acid prototrophy is the ancestral state for free-living bacteria. This aligns with the expectation that essential metabolism includes the core biosynthetic pathways.
The high conservation (17/18 pathways at 100% within sample) indicates these biosynthetic routes represent a minimal metabolic repertoire required for independent growth. Organisms lacking these pathways (auxotrophs) depend on external nutrient sources or live in symbiotic/commensal relationships.
Ecological Interpretation of DvH Serine Auxotrophy
Desulfovibrio vulgaris is known for:
- Anaerobic respiration using sulfate as terminal electron acceptor
- Organic acid utilization (lactate, formate, pyruvate)
- Niche: Anaerobic sediments, biofilms, intestinal tracts
The apparent serine auxotrophy makes ecological sense:
1. These environments are rich in amino acids from protein degradation
2. Streamlining metabolism by losing biosynthetic capacity could be advantageous when nutrients are abundant
3. This may represent genomic economy - losing genes that are energetically costly when the product is freely available
Alternative Explanations
GapMind detection limitations:
1. DvH may use a non-canonical serine biosynthesis pathway not in GapMind's reference database
2. The pathway may be present but use divergent enzymes that fall below GapMind's homology threshold
3. Genes may be present but unannotated in the genome assembly
Future validation: Experimental testing of DvH growth on serine-free minimal media would confirm auxotrophy.
Hypothesis Outcome
Original H1 (Revised version): "A core set of metabolic pathways is universally complete across bacteria"
Result: Partially supported with significant caveats
✅ Supported:
- High pathway conservation observed (17/18 AA pathways, most carbon sources)
- Minimal metabolic repertoire identified for amino acid biosynthesis
- Central catabolic pathways widely shared
❌ Not supported:
- NO pathways are truly universal (100%) even in this small sample
- Organism-specific gaps exist (DvH serine)
- Analysis limited to 7 organisms (not pan-bacterial)
Conclusion: The data reveal near-universal rather than strictly universal metabolic pathway completeness. Even among organisms with experimental essential gene data, some metabolic diversity exists, likely reflecting ecological adaptation and nutrient availability.
Future Directions
Immediate Follow-ups
- Literature review: Is D. vulgaris serine auxotrophy documented experimentally?
- Check lower-confidence GapMind predictions: Does DvH have serine pathway with "steps_missing" status?
- Expand organism mapping: Add more FB organisms to increase sample size
Alternative Approaches
- eggNOG pathway analysis: Use EC annotations → KEGG pathways for all 45 FB organisms (including E. coli)
- Hybrid approach: Combine GapMind (pathway-level) with eggNOG (gene-level) for comprehensive coverage
- Essential gene → pathway linkage: Map essential genes directly to metabolic pathways rather than relying on GapMind
Broader Questions
- Pathway essentiality vs completeness: Are "complete" pathways actually essential for viability?
- Conditional essentiality: Which pathways are essential only under specific growth conditions?
- Phylogenetic patterns: Do pathway gaps cluster by phylogeny or ecology?
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
gapmind_pathways, genome |
Pathway completeness predictions (305M predictions, 293K genomes) |
projects/essential_genome |
essential_families.tsv |
Universally essential gene families (859 families, 45 organisms) |
Generated Data
| File | Rows | Description |
|---|---|---|
data/pathway_completeness.tsv |
80 | All pathway completeness across 7 organisms |
data/aa_pathway_completeness.tsv |
18 | Amino acid biosynthesis pathways |
data/carbon_pathway_completeness.tsv |
62 | Carbon source utilization pathways |
data/gapmind_fb_predictions.tsv |
7,389 | Raw GapMind predictions for 7 organisms |
data/fb_genome_mapping_manual.tsv |
8 | FB organism → genome ID mappings |
References
- Price MN et al. (2020). "Filling gaps in bacterial amino acid biosynthesis pathways with high-throughput genetics." PLoS Genet 16(1):e1008594. DOI: 10.1371/journal.pgen.1008594. PMID: 31951614 (GapMind methodology)
- Heidelberg JF et al. (2004). "The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough." Nat Biotechnol 22:554-559. DOI: 10.1038/nbt959. PMID: 15077118
- Price MN et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557:503-509. DOI: 10.1038/s41586-018-0124-0. PMID: 29769716 (Fitness Browser)
- Parks DH et al. (2022). "GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy." Nucleic Acids Res 50:D785-D794. DOI: 10.1093/nar/gkab776. PMID: 34520557 (GTDB/pangenome)
Data Collections
Derived Data
This project builds on processed data from other projects.
Used By
Data from this project is used by other projects.
Review
Summary
This pilot study successfully demonstrates a pathway-level approach to characterizing the essential metabolome across bacteria, finding near-universal conservation of amino acid biosynthesis pathways (17/18 pathways present in all 7 organisms analyzed). The project exhibits excellent scientific practices including transparent limitation reporting, thoughtful methodological pivoting when the original EC→reaction approach proved infeasible, and proper data provenance. The key finding—that Desulfovibrio vulgaris lacks serine biosynthesis while maintaining complete pathways for 17 other amino acids—is well-supported and receives appropriate ecological interpretation. The analysis is limited to 7 organisms (not the intended 45) due to incomplete GapMind coverage, particularly E. coli's complete absence from the dataset, but this limitation is honestly acknowledged throughout the documentation.
Methodology
Research Question: Clearly stated and testable. The original question ("Which biochemical reactions are universally essential?") appropriately evolved to "Which metabolic pathways are universally complete?" when biochemistry database limitations were discovered. The revision history in RESEARCH_PLAN.md transparently documents this evolution (v1→v2→v3).
Approach: Sound and well-justified. The project:
1. References 859 universally essential gene families from the essential_genome project with proper attribution
2. Maps 8 Fitness Browser organisms to GTDB genome IDs (manual mapping documented in data/fb_genome_mapping_manual.tsv)
3. Queries GapMind pathway completeness predictions from kbase_ke_pangenome (305M predictions)
4. Analyzes pathway conservation patterns across amino acid biosynthesis and carbon source utilization
Data Sources: Clearly identified with appropriate attribution and scale documentation. All source tables are explicitly referenced with row counts and provenance information in both README and REPORT.
Reproducibility Gaps:
- ✅ Notebook outputs: Excellent. The main analysis notebook (02_gapmind_pathway_analysis.ipynb) contains complete outputs for all 22 cells, including text results, data summaries, and the embedded pathway completeness figure. The exploratory notebook (01_extract_essential_reactions.ipynb) also has complete outputs documenting the EC→reaction investigation.
- ✅ Figures: One comprehensive figure (pathway_completeness.png, 263 KB) effectively visualizes the key findings with dual panels for amino acid pathways and carbon sources. For a pilot study with limited scope, this is adequate.
- ✅ Dependencies: requirements.txt is present with appropriate packages (pandas, pyspark, scipy, matplotlib, seaborn, networkx) and version constraints.
- ⚠️ Reproduction guide: The README includes a complete Reproduction section (lines 83-103) documenting prerequisites, steps, expected runtimes, and Spark Connect requirements. This satisfies reproducibility requirements.
- ✅ Separation of concerns: The README clearly indicates that GapMind analysis is the completed approach, while noting that 01_extract_essential_reactions.ipynb represents exploratory work on the abandoned EC→reaction method.
Code Quality
SQL Queries: All Spark SQL queries in the main analysis notebook are correct and efficient:
- Proper use of IN clause for filtering to 8 genome IDs
- Appropriate column selection from gapmind_pathways table
- No use of inefficient patterns like LIKE when exact equality is available
- Results are filtered in Spark before .toPandas() conversion (good practice per docs/pitfalls.md)
Pitfall Awareness: The analysis correctly handles the GapMind multi-row structure documented in docs/pitfalls.md (lines 692-736). The notebook uses .groupby().agg({'genome_id': 'nunique'}) to count distinct genomes per pathway, which properly handles multiple rows per genome-pathway pair. While the pitfall documentation recommends taking MAX score per genome-pathway, the notebook's approach of filtering to complete/likely_complete categories before counting is equally valid.
Statistical Methods: Appropriate for pilot scope. The analysis uses descriptive statistics (counts, percentages) rather than inferential tests, which is suitable for n=7 organisms and exploratory objectives. No concerns about statistical rigor.
Notebook Organization: The main analysis notebook is well-structured with clear logical flow: Setup → Load organism list → Load genome mapping → Extract GapMind predictions → Analyze completeness → Visualize → Save results. Markdown cells provide context for each step, and print statements give clear progress indicators.
Code Issues: None identified. Variable names are descriptive, pandas operations are appropriate, and the code successfully handles the discovery that only 7 of 8 mapped organisms have GapMind data (Keio/E. coli missing).
Findings Assessment
Amino Acid Biosynthesis Conservation: The claim that "17 of 18 amino acid biosynthesis pathways are present in all 7 organisms" is directly supported by data/aa_pathway_completeness.tsv, which shows 17 pathways with n_complete=7 and serine with n_complete=6. This is accurately reported in README, REPORT, and visualized in the figure.
DvH Serine Auxotrophy: The identification of Desulfovibrio vulgaris as lacking serine biosynthesis is supported by the pathway completeness data. The REPORT provides thoughtful ecological interpretation (anaerobic sulfate-reducer in organic-rich environments) and appropriately considers alternative explanations (non-canonical pathway, GapMind detection limits). The interpretation is scientifically sound and avoids overstatement.
Carbon Source Conservation: The finding that TCA intermediates (fumarate, succinate), fermentation products (acetate, propionate, L-lactate), and amino acid catabolism pathways are widely conserved (7/7 organisms) is supported by data/carbon_pathway_completeness.tsv.
Limitations Acknowledged: Excellent. The REPORT includes a comprehensive Limitations section (lines 126-146) that honestly addresses:
- Small sample size (7 organisms, not 45)
- Limited phylogenetic diversity (mostly Proteobacteria)
- Cannot generalize to pan-bacterial scale
- GapMind coverage gaps (E. coli completely absent)
- Computational predictions vs experimental validation
- RB-TnSeq rich media effects on apparent essentiality
The README and RESEARCH_PLAN also clearly state this is a "pilot study" due to coverage limitations. This transparency reflects excellent scientific integrity.
Incomplete Analysis: The README "Project Structure" section (lines 38-60) accurately reflects the delivered outputs for the GapMind pathway analysis approach. The original plan's EC→reaction outputs are appropriately marked as "not completed" with explanation of why the approach was abandoned.
Visualizations: The single figure (pathway_completeness.png) is publication-quality with clear axis labels, appropriate reference lines at 90% and 100% completeness thresholds, and effective dual-panel layout. The figure is properly referenced in the REPORT and effectively supports the key findings.
Suggestions
High-Priority (Addressing Gaps)
- Verify notebook output cell rendering: While the notebooks contain outputs in the
.ipynbfile, the Read tool indicated that cell 18 of notebook 02 had outputs "too large to include." Verify that the embedded figure in that cell renders properly when opened in JupyterLab. If the output is truncated, consider re-running the cell withplt.show()to ensure the figure is saved to the filesystem but the notebook output is a smaller reference.
Medium-Priority (Enhancing Impact)
- Expand organism coverage to strengthen generalizability: The analysis is currently limited to 7 organisms. Two approaches could increase coverage:
- Option A: Map additional Fitness Browser organisms to genome IDs. The current 8 mapped organisms represent only 17.8% of the 45 FB organisms with essential gene data.
- Option B: Implement the eggNOG → KEGG pathway approach mentioned in REPORT "Future Directions" (lines 183-186). This would enable inclusion of all 45 organisms including E. coli, providing a more robust pan-bacterial analysis.
-
Impact: Would transform this from a pilot study to a comprehensive analysis supporting stronger conclusions about universal metabolic requirements.
-
Validate DvH serine auxotrophy via literature search: The REPORT suggests checking PubMed for experimental evidence of serine requirement in Desulfovibrio vulgaris (line 179). Use the
/literature-reviewskill to search for studies of DvH amino acid requirements or serine biosynthesis. This would either confirm the computational prediction or identify alternative pathways that GapMind may have missed.
Low-Priority (Optional Enhancements)
-
Add phylogenetic context visualization: The 7 organisms represent limited phylogenetic diversity (6 Proteobacteria + 1 Deltaproteobacterium). A phylogenetic tree showing their relationships with pathway completeness overlaid would help readers assess whether the patterns reflect phylogenetic constraint or functional convergence. This is optional given the pilot scope.
-
Document genome mapping methodology: The file
data/fb_genome_mapping_manual.tsvcontains mappings but doesn't document how they were created. Adding a brief note in README or adata/README.mdexplaining the mapping process (e.g., "Mappings created by matching FB organism names to NCBI RefSeq accessions via NCBI Taxonomy, then queryingkbase_ke_pangenome.genomefor corresponding GTDB genome IDs") would improve reproducibility for future organism additions. -
Consider adding pathway-organism heatmap: While the current figure effectively shows pathway completeness, a heatmap showing which organism lacks which pathway would make the DvH serine gap more visually apparent. This is optional as the current figure adequately communicates the main findings.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Pathway Completeness
Notebooks
Data Files
| Filename | Size |
|---|---|
aa_pathway_completeness.tsv |
0.3 KB |
carbon_pathway_completeness.tsv |
1.4 KB |
essential_genome_families.tsv |
2800.3 KB |
fb_genome_mapping_manual.tsv |
0.6 KB |
gapmind_aa_pathway_summary.tsv |
0.6 KB |
gapmind_fb_predictions.tsv |
630.8 KB |
pathway_completeness.tsv |
1.7 KB |