Pan-bacterial Fitness Modules via Independent Component Analysis
CompletedResearch Question
Can we decompose RB-TnSeq fitness compendia into latent functional modules via robust ICA, align them across organisms using orthology, and use module context to predict gene function?
Research Plan
Hypothesis
Robust ICA applied to gene-fitness matrices will identify stable, biologically coherent modules of co-regulated genes. These modules can be aligned across organisms using ortholog fingerprints to reveal conserved fitness regulons, and module membership can be used to predict function for unannotated genes.
Approach
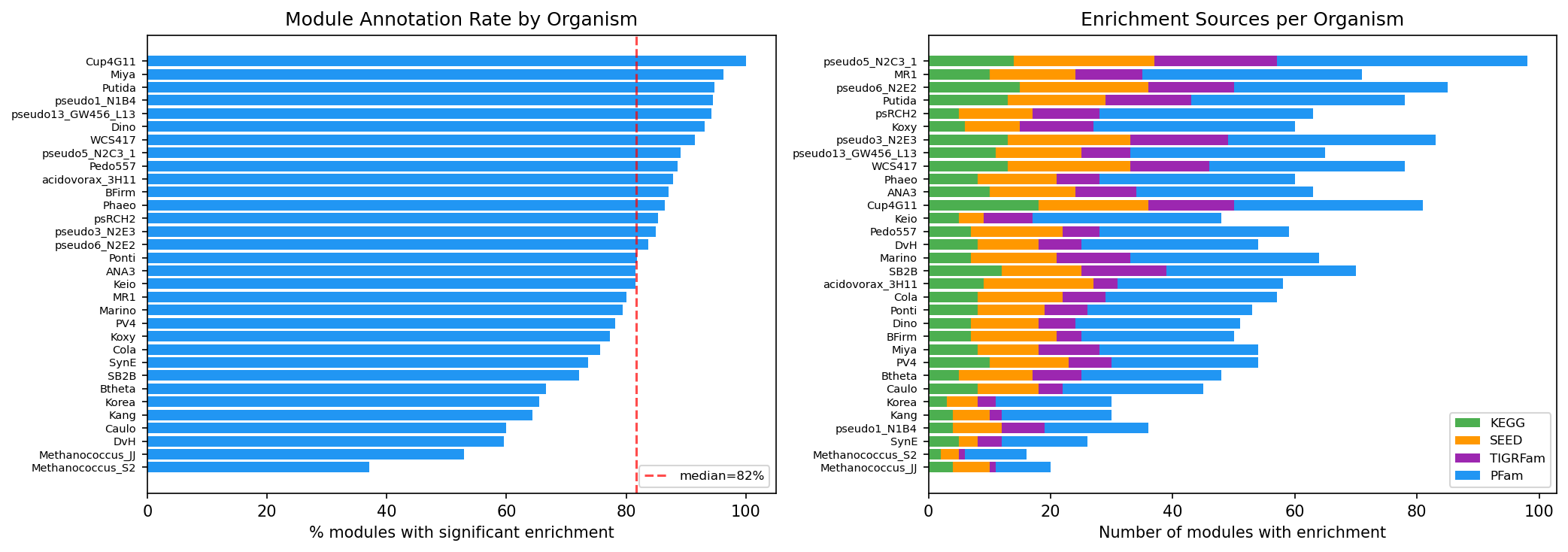
- Explore & select -- Surveyed all 48 organisms; selected 32 with >=100 experiments
- Extract matrices -- Built gene x experiment fitness matrices from Spark (2,531-6,384 genes x 104-757 experiments)
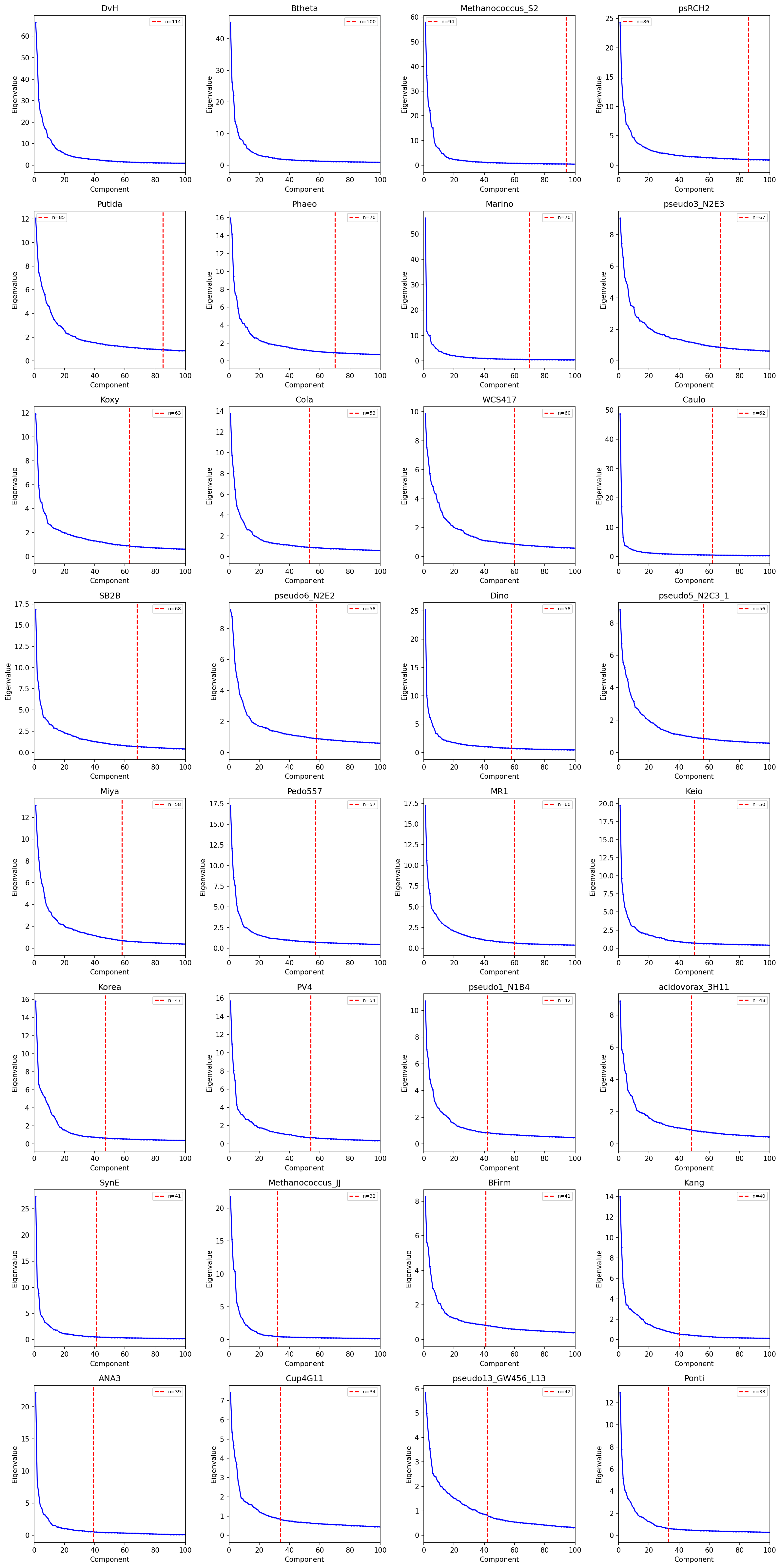
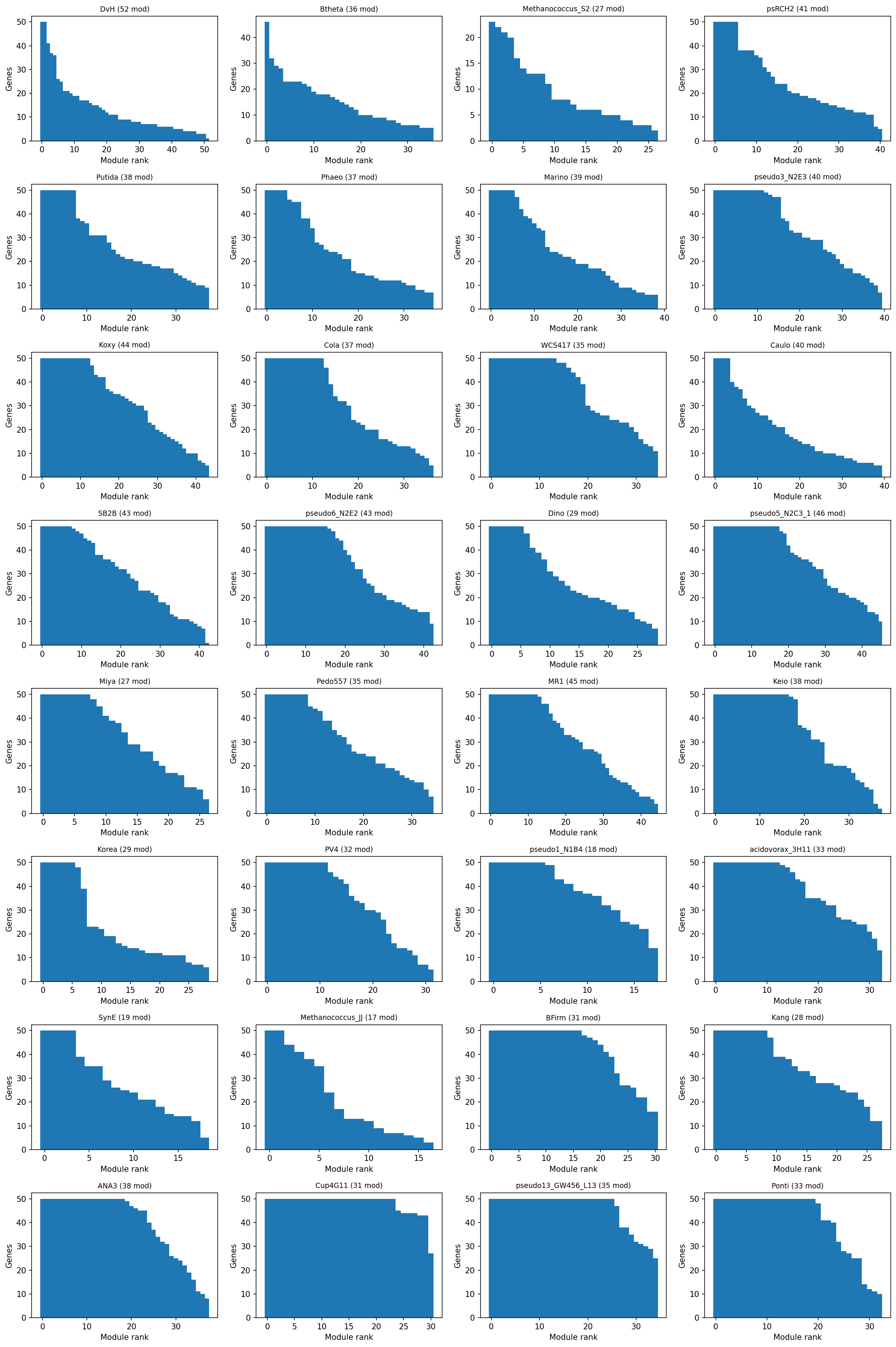
- Robust ICA -- 30-50x FastICA runs -> DBSCAN clustering -> stable modules (5-50 genes each)
- Annotate modules -- KEGG, SEED, TIGRFam, PFam enrichment (Fisher exact + BH FDR); map to conditions
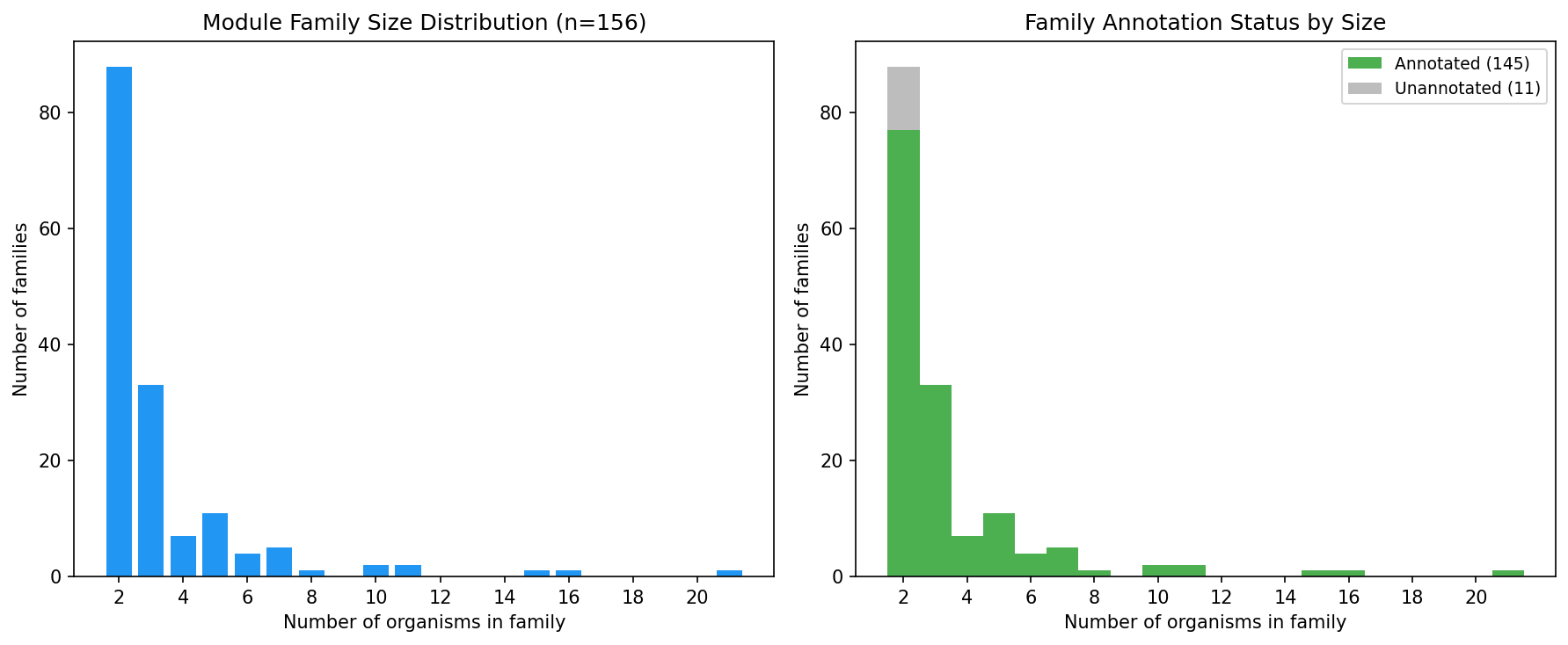
- Cross-organism alignment -- 1.15M BBH pairs -> 13,402 ortholog groups -> module family fingerprints -> 156 families
- Predict function -- Module + family context -> 6,691 function predictions for hypothetical proteins
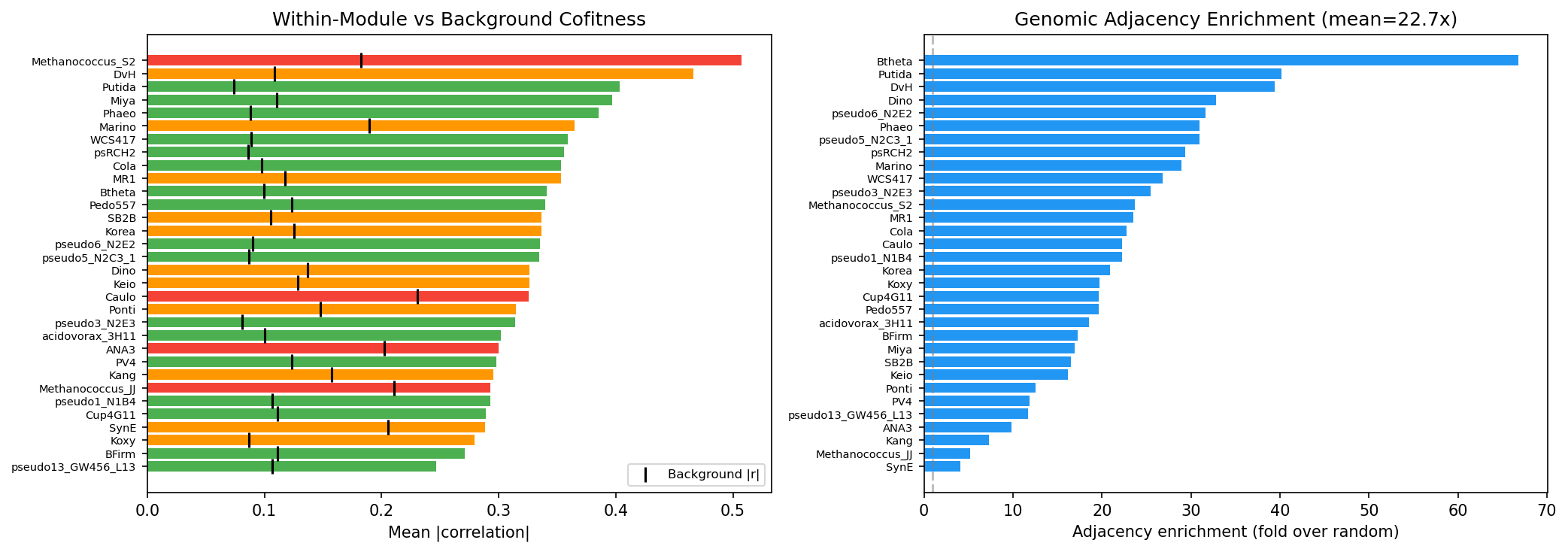
- Validate -- Within-module cofitness (94.2% enriched) and genomic adjacency (22.7x enrichment)
Revision History
- v1 (2026-02): Migrated from README.md
Overview
This project applies robust Independent Component Analysis (ICA) to gene-fitness matrices from the Fitness Browser, decomposing them into latent functional modules of co-regulated genes. Modules are aligned across 32 bacterial organisms using ortholog fingerprints to reveal conserved fitness regulons, and module membership is used to predict biological process context for unannotated genes. The approach complements sequence-based methods by capturing process-level co-regulation rather than specific molecular function.
Key Findings
-
The strict membership threshold (|weight| >= 0.3, max 50 genes) was critical. The initial D'Agostino K-squared approach gave 100-280 genes per module with weak cofitness signal (59% enriched, 1-17x correlation). After switching to absolute weight thresholds, modules became biologically coherent (94% enriched, 2.8x correlation enrichment).
-
Adding PFam domains and lowering the enrichment overlap threshold from 3 to 2 increased module annotation rate from 8% to 80% (92 to 890 modules), unlocking 7.6x more function predictions. PFam provides the broadest annotation coverage; KEGG KOs are too gene-specific for module-level enrichment.
-
Module-ICA is complementary to sequence-based methods: it excels at identifying co-regulated gene groups (biological process modules) but should not be used for predicting specific molecular functions, where ortholog transfer is far superior.
Results
ICA Decomposition (32 organisms)
- 1,116 stable modules across 32 organisms (all with >=100 experiments)
- Module sizes: median 7-50 genes per module (biologically correct range)
- 94.2% of modules show significantly elevated within-module cofitness (Mann-Whitney U, p < 0.05)
- Within-module mean |r| = 0.34 vs background |r| = 0.12 (2.8x enrichment)
- 22.7x genomic adjacency enrichment -- module genes are co-located in operons
Provenance:
notebooks/03_ica_modules.ipynb-- Robust ICA decomposition, PCA component selection, module extraction, and cofitness validation.
Benchmarking (NB07)
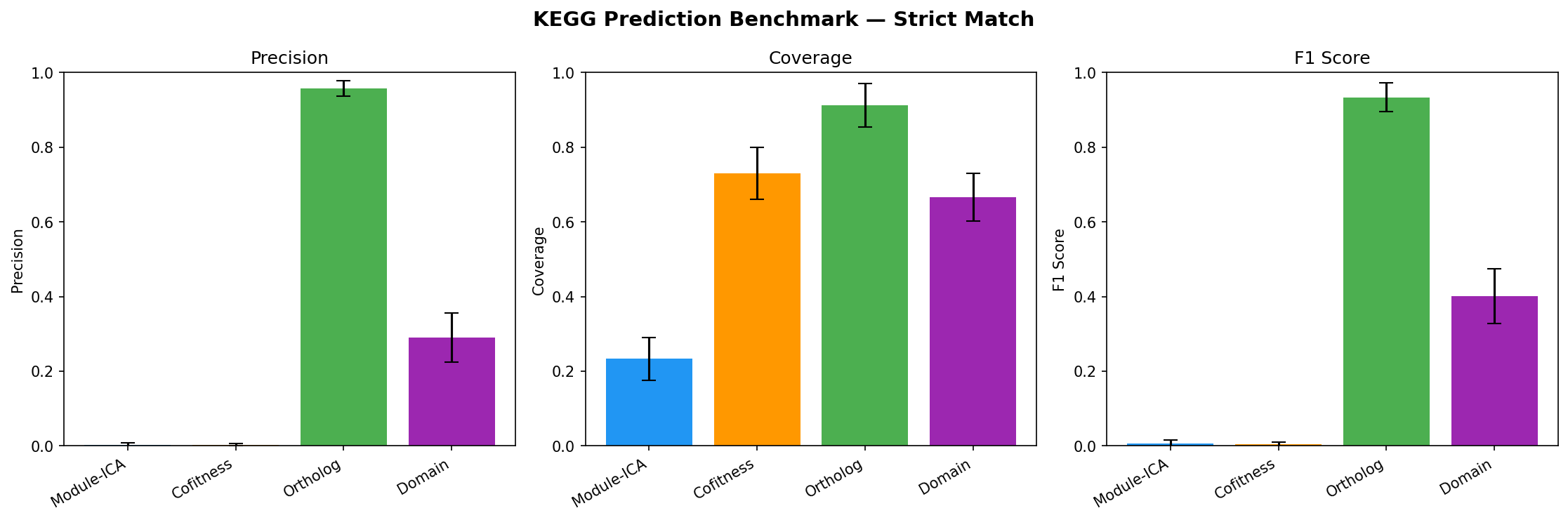
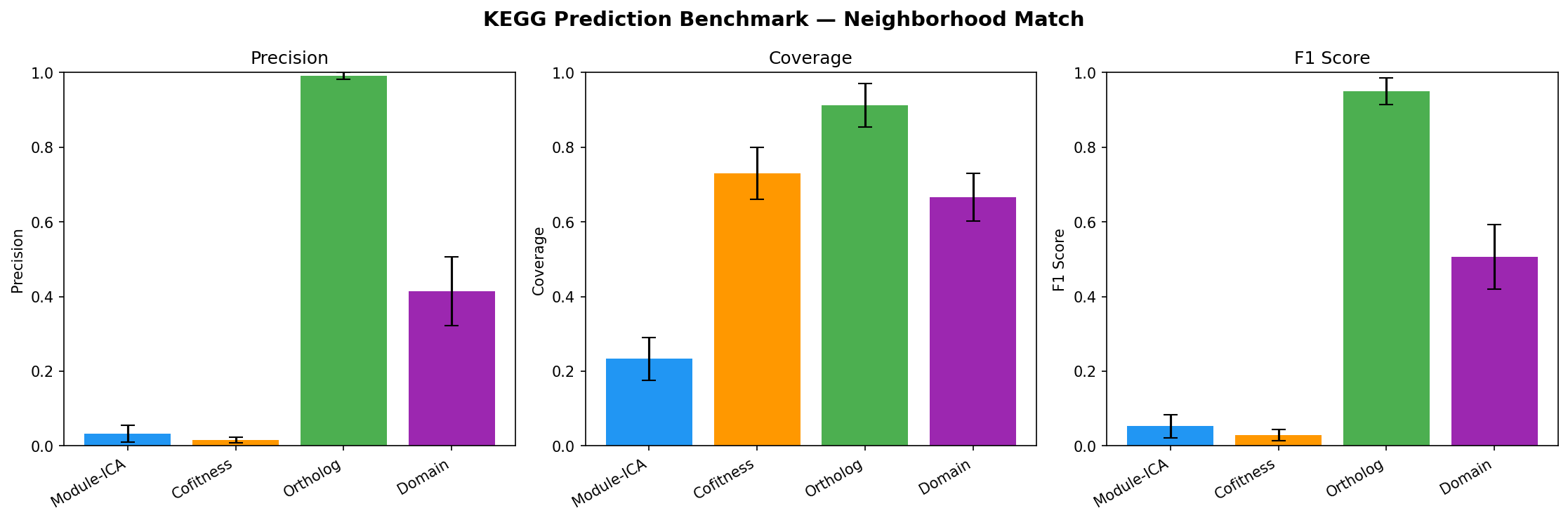
Held-out evaluation: 20% of KEGG-annotated genes withheld, 4 methods predict KO groups.
| Method | Precision (strict) | Coverage | F1 |
|---|---|---|---|
| Ortholog transfer | 95.8% | 91.2% | 0.934 |
| Domain-based | 29.1% | 66.6% | 0.401 |
| Module-ICA | <1% | 23.3% | -- |
| Cofitness voting | <1% | 73.0% | -- |
Module-ICA and cofitness show near-zero strict KO precision because KEGG KO groups are gene-level assignments (~1.2 genes per unique KO). A module with 20 annotated members typically has 20 different KOs. Modules capture process-level co-regulation (validated by 94.2% cofitness enrichment and 22.7x adjacency enrichment), not specific molecular function. Function predictions should be interpreted as biological process context, not exact KO assignments.
Provenance:
notebooks/07_benchmarking.ipynb-- Held-out evaluation of function prediction methods;notebooks/03_ica_modules.ipynb-- cofitness validation.
Cross-Organism Alignment
- 1.15M BBH pairs across 32 organisms -> 13,402 ortholog groups
- 156 module families spanning 2+ organisms (28 spanning 5+, 7 spanning 10+, 1 spanning 21)
- 145 annotated families with consensus functional labels (93%)
- Largest family spans 21 organisms -- a pan-bacterial fitness module
Provenance:
notebooks/05_cross_organism_alignment.ipynb-- Ortholog fingerprinting and module family construction.
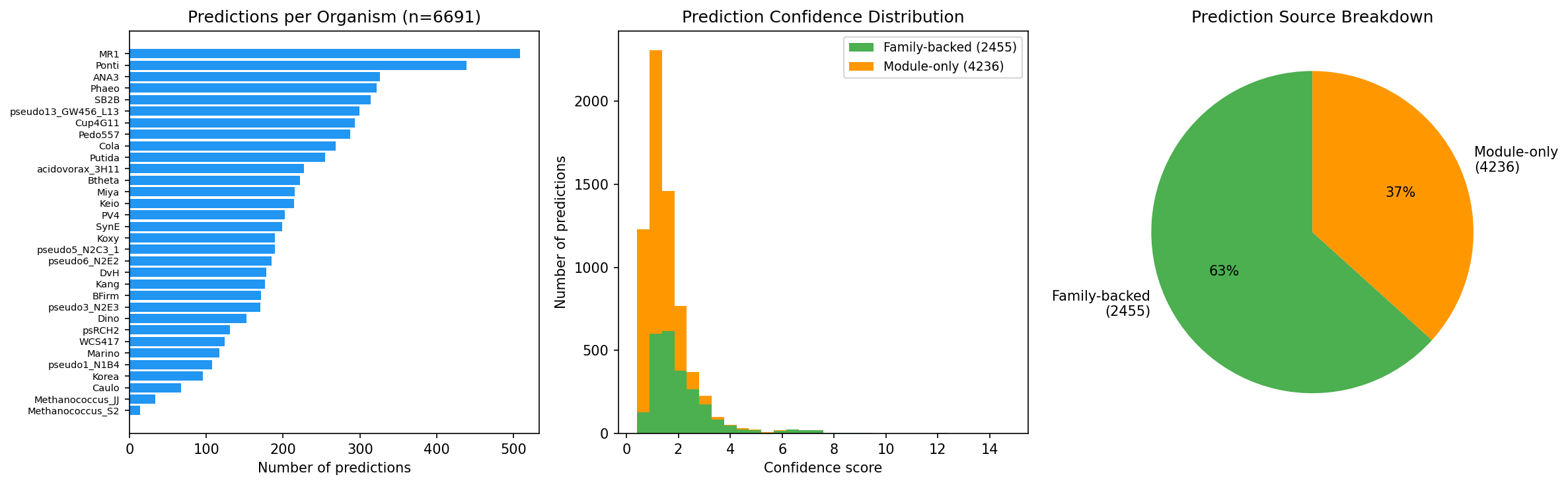
Function Prediction
- 6,691 function predictions for hypothetical proteins across all 32 organisms
- 2,455 family-backed (37%) -- supported by cross-organism conservation
- 4,236 module-only predictions
- Predictions backed by module enrichment (KEGG, SEED, TIGRFam, PFam)
Provenance:
notebooks/04_module_annotation.ipynb-- Functional enrichment analysis;notebooks/06_function_prediction.ipynb-- Function prediction for hypothetical proteins.
Interpretation
Module-ICA provides a complementary layer of functional annotation that captures biological process co-regulation rather than molecular function identity. Ortholog transfer remains the gold standard for gene-level function prediction (95.8% precision), but Module-ICA fills a different niche: identifying which biological processes an uncharacterized gene participates in. The 6,691 function predictions for hypothetical proteins should be interpreted as "involved in [biological process]" rather than "has function [specific KO]."
The cross-organism alignment demonstrates that conserved fitness regulons exist across diverse bacterial phyla. The largest module family spanning 21 organisms provides evidence for deeply conserved co-regulation programs.
Literature Context
-
ICA for gene module detection: Sastry et al. (2019) demonstrated that the E. coli transcriptome consists largely of independently regulated modules recoverable by ICA. Our cofitness-based approach extends this framework from transcriptomic to fitness data across 32 organisms, confirming that ICA captures biologically coherent co-regulation structure in phenotypic compendia as well. Sastry AV et al. "The Escherichia coli transcriptome mostly consists of independently regulated modules." Nat Commun 10, 5536 (2019). PMID: 31767856
-
Cofitness as a proxy for co-regulation: Price et al. (2018) showed that mutant fitness data across thousands of conditions reveals gene function at scale. Our use of cofitness (within-module correlation) as the primary validation metric builds on their finding that genes with correlated fitness profiles share biological function. Price MN et al. "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557, 503--509 (2018). PMID: 29769716
Limitations
- Module-ICA has near-zero precision for predicting specific KEGG KO assignments; it captures process-level, not gene-level, function
- Organisms with fewer than ~100 experiments produce weaker modules (e.g., Caulo with 198 experiments showed only 2.9x correlation enrichment)
- The 40% component cap (components <= 40% of experiments) is necessary to avoid FastICA convergence failures but may miss some modules in low-experiment organisms
- PFam-based annotations, while providing the best coverage, are at the domain level and may overcount functional associations
Future Directions
- Extend to additional organisms as new RB-TnSeq datasets become available
- Integrate module predictions with pangenome gene classifications (core/auxiliary/singleton)
- Apply module-based analysis to specific biological questions (e.g., which modules are enriched in accessory genes?)
- Develop a web interface for browsing module families and function predictions
Data
Sources
| Database | Tables | Description |
|---|---|---|
kescience_fitnessbrowser |
genefitness, gene, exps, ortholog |
RB-TnSeq fitness scores, gene metadata, experiment conditions, and ortholog mappings from the Fitness Browser |
kbase_ke_pangenome |
gene_cluster |
Pangenome gene cluster assignments for cross-organism alignment |
Generated Data
| Path | Description |
|---|---|
data/matrices/ |
Per-organism gene-fitness matrices extracted from Spark (NB02) |
data/annotations/ |
Gene and experiment metadata, functional annotations (KEGG, SEED, TIGRFam, PFam) |
data/orthologs/ |
Bidirectional best-hit (BBH) ortholog pairs and ortholog groups |
data/modules/ |
ICA module definitions, gene weights, and per-organism ICA parameters (32 *_ica_params.json files) |
data/module_families/ |
Cross-organism module families constructed via ortholog fingerprints |
data/predictions/ |
Function predictions for hypothetical proteins and benchmark results |
References
- Borchert AJ et al. (2019). "Proteome and transcriptome analysis of Pseudomonas putida KT2440 using independent component analysis." mBio.
- Price MN et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature.
- Hyvarinen A & Oja E (2000). "Independent component analysis: algorithms and applications." Neural Networks.
- Sastry AV et al. (2019). "The Escherichia coli transcriptome mostly consists of independently regulated modules." Nat Commun.
Discoveries
Robust ICA (30-50 FastICA runs + DBSCAN clustering) consistently finds 17-52 stable modules per organism across 32 bacteria. 94.2% of modules show significantly elevated within-module cofitness (Mann-Whitney U, p < 0.05; mean |r| = 0.34 vs background 0.12). Genomic adjacency enrichment averages 22.7
Read more →The initial D'Agostino K² normality-based thresholding gave 100-280 genes per module — biologically meaningless. The ICA components themselves were fine; the problem was deciding which genes "belong" to each module. Switching to an absolute weight threshold (|Pearson r| ≥ 0.3 with module profile, ma
Read more →Caulo (198 experiments) showed the weakest signal in the pilot (2.9x correlation enrichment vs 31x for DvH), but organisms down to 104 experiments (Ponti) still produced stable modules. The key is capping components at 40% of experiments — higher ratios cause FastICA convergence failures and extreme
Read more →Fitness Browser schema documentation has inaccuracies
February 2026Several table schemas differ from the documented schema:
- keggmember uses keggOrg/keggId (not orgId/locusId) — must join through besthitkegg
- kgroupec uses ecnum (not ec)
- seedclass has orgId, locusId, type, num (not subsystem/category hierarchy)
- fitbyexp_* tables are lo
Spark is accessible from CLI via berdl_notebook_utils
February 2026from berdl_notebook_utils.setup_spark_session import get_spark_session works from regular Python scripts on JupyterHub — not just notebook kernels. This enables running full analysis pipelines from the command line without jupyter nbconvert. The auto-import in `/configs/ipython_startup/00-notebo
Pan-bacterial module families exist across diverse phyla
February 2026Cross-organism module alignment using BBH ortholog fingerprints revealed 156 module families spanning 2+ organisms. The largest family spans 21 of 32 organisms across Proteobacteria, Bacteroidetes, Firmicutes, and Archaea — evidence of deeply conserved fitness regulons. 28 families span 5+ organisms
Read more →Using BBH pairs from 5 organisms (10K pairs, 1,861 OGs) vs all 32 organisms (1.15M pairs, 13,402 OGs) produced radically different results:
- Module families: 27 → 156 (6x)
- Families spanning 5+ orgs: 0 → 28
- Family-backed predictions: 31 (4%) → 493 (56%)
Lesson: always extract orthologs for ALL
Read more →The initial enrichment pipeline (KEGG + SEED + TIGRFam, min_annotated=3) annotated only 8.2% of modules (92/1,116). Root cause: KEGG KO groups average ~1.2 genes per term, so modules with 5-50 members almost never have 3+ genes sharing the same KO. Adding PFam domains (which have 814 terms with 2+ g
Read more →Held-out KO prediction benchmark across 32 organisms showed ortholog transfer dominates at gene-level function prediction (95.8% precision, 91.2% coverage), while Module-ICA has <1% precision at the KO level. This is expected and informative, not a failure: modules capture **process-level co-regulat
Read more →The min_annotated parameter (minimum overlapping genes to test for enrichment) must be calibrated to the annotation database's granularity. For gene-specific annotations (KEGG KOs: ~1 gene/term), min_annotated=3 eliminates nearly all tests. For domain-level annotations (PFam: many genes share domain
Read more →Data Collections
Used By
Data from this project is used by other projects.
Pan-Bacterial Metal Fitness Atlas
View project →
Gene Conservation, Fitness, and the Architecture of Bacterial Genomes
View project →
Co-fitness Predicts Co-inheritance in Bacterial Pangenomes
View project →
Field vs Lab Gene Importance in Desulfovibrio vulgaris Hildenborough
View project →
The Pan-Bacterial Essential Genome
View project →
Fitness Modules x Pangenome Conservation
View project →
Truly Dark Genes — What Remains Unknown After Modern Annotation?
View project →
Review
Summary
This is an impressive, well-executed computational biology project that decomposes RB-TnSeq fitness compendia from 32 bacterial organisms into 1,116 stable ICA modules, aligns them across organisms via ortholog fingerprints into 156 module families, and generates 6,691 function predictions for hypothetical proteins. The project demonstrates strong software engineering and scientific practices: a clean 7-notebook pipeline with consistent structure, reusable source modules (ica_pipeline.py, run_benchmark.py), idempotent caching at every stage, four annotation databases, a formal 4-method benchmark with dual-granularity evaluation, and honest reporting of method limitations. The README is exemplary — complete reproduction guide, clear Spark/local separation, data source table, and well-contextualized results. Validation is compelling: 94.2% of modules show significantly elevated within-module cofitness and 22.7× genomic adjacency enrichment. The main areas for improvement are: (1) NB01–02 lack saved cell outputs, making the early pipeline opaque to readers without Spark access; (2) the cofitness validation could be strengthened with multiple-testing correction across 1,114 modules; and (3) predicted functions are often domain-family labels (e.g., "DEAD", "ABC_tran") rather than biological process descriptions, creating a semantic gap with the stated goal of process-level prediction.
Methodology
Research question: Clearly stated, testable, and well-motivated — "Can we decompose RB-TnSeq fitness compendia into latent functional modules via robust ICA, align them across organisms using orthology, and use module context to predict gene function?" The three-part question maps directly to notebooks 03–06, with NB07 providing formal evaluation.
Approach: Sound and methodologically rigorous. The pipeline follows Borchert et al. (2019) with meaningful extensions to a pan-bacterial framework:
- Marchenko-Pastur eigenvalue thresholding for component selection (capped at 40% of experiments, max 80)
- 30–50× FastICA restarts with DBSCAN stability filtering (eps=0.15)
- Fisher exact enrichment with BH FDR correction across four annotation databases
- Ortholog-group fingerprinting via connected components on 1.15M BBH pairs
- Hierarchical clustering into module families (cosine distance, average linkage, t=0.7)
- Dual-level benchmarking (strict KO match + neighborhood set overlap) on 20% held-out genes
The key methodological insight — that absolute weight thresholds (|r| ≥ 0.3, max 50 genes) dramatically outperform D'Agostino K² thresholding for ICA membership — is well-documented and represents a valuable contribution.
Data sources: Clearly identified in the README with a table mapping 10 kescience_fitnessbrowser tables to their use. SQL queries throughout correctly follow the documented join paths and type-casting requirements.
Reproducibility:
- Reproduction guide: The README includes a thorough ## Reproduction section with prerequisites, step-by-step nbconvert commands, clear Spark/local separation, runtime estimates (30–80 min per organism for ICA, ~1 min from cache), and a standalone benchmark command.
- Notebook outputs: NB03–07 all have saved outputs (text tables, progress logs, embedded figures). NB01–02 have zero saved outputs across all code cells. This is documented with a header note, but means readers cannot see organism selection rationale or matrix shapes without JupyterHub access. The "Execution Summary" markdown cells partially mitigate this.
- Figures: 8 substantive PNG files covering all major stages: PCA eigenvalues, module size distributions, enrichment summary, module families, prediction summary, validation summary, and both benchmark charts.
- Dependencies: requirements.txt with 7 packages and minimum version constraints.
- Caching: All notebooks implement file-existence checks. All 32 organisms have ICA parameter JSON files recording exact hyperparameters.
Code Quality
SQL correctness: All queries are correct and demonstrate awareness of documented pitfalls:
- String-to-numeric casting applied consistently (CAST(cor12 AS FLOAT), CAST(fit AS FLOAT), CAST(begin AS INT))
- KEGG join path in NB04 correctly routes through besthitkegg with composite keys (bk.keggOrg = km.keggOrg AND bk.keggId = km.keggId)
- SEED annotations correctly use seedannotation.seed_desc, avoiding the misleading seedclass table
- Experiment QC filtering (cor12 >= 0.1) and gene missingness filtering (≤50% missing) are reasonable
Pitfall awareness: Outstanding — the project contributed 7 fitness-browser-specific pitfalls to docs/pitfalls.md (all tagged [fitness_modules]):
- ✅ Cosine distance floating-point fix (np.clip, ica_pipeline.py line 146)
- ✅ KEGG join path through besthitkegg
- ✅ seedclass vs seedannotation correction
- ✅ D'Agostino K² thresholding failure documented
- ✅ Ortholog scope must match analysis scope
- ✅ ICA component ratio capped at 40%
- ✅ Enrichment min_annotated granularity mismatch
Statistical methods: Appropriate throughout:
- Fisher exact test with BH FDR for enrichment — contingency table correctly constructed with alternative='greater'
- Mann-Whitney U for cofitness validation — appropriate non-parametric test
- DBSCAN for component clustering — good choice for unknown cluster count
- One concern: cofitness validation tests 1,114 modules at p < 0.05 without multiple-testing correction. At 5% FDR, ~56 modules could be falsely enriched. The 94.2% rate is robust enough that this doesn't change the conclusion, but the omission is methodologically inconsistent with the careful FDR correction applied in NB04.
ica_pipeline.py (295 lines): Clean, well-documented module with proper docstrings, edge-case handling (zero-norm vectors, empty arrays), and correct sign-alignment via dot-product reference.
run_benchmark.py (749 lines): Comprehensive standalone script with vectorized correlation, proper train/test separation, and CSV output for all validation results. Good use of matplotlib.use("Agg") for headless execution.
Minor issues:
1. ortholog_predict in run_benchmark.py (line 196) takes the first BBH hit with a break, ignoring the ratio column. Sorting by ratio descending first would use the highest-quality ortholog.
2. The fitbyexp_* extraction function in NB02 attempts to parse these tables as wide format, but the documented pitfall notes they are long format. The function fails silently and the correct genefitness fallback triggers — harmless but dead code.
3. Within-module correlation extraction in run_benchmark.py (lines 604–607) uses nested Python loops rather than vectorized numpy indexing, inconsistent with the vectorized style elsewhere.
Notebook organization: Consistent pattern across all 7 notebooks: markdown header → imports/setup → numbered sections → visualization → summary. Caching status always reported. Spark/local separation clearly marked.
Findings Assessment
Conclusions well-supported by data:
- 1,116 stable modules — verified by NB03 cell 8 (1,117 stable, 1,116 with members) and 32 JSON parameter files
- 94.2% cofitness enrichment — 1,049/1,114 modules, with 17/32 organisms at 100% and all but 3 above 75%
- 22.7× genomic adjacency enrichment — per-organism range 4.1× (SynE) to 66.8× (Btheta)
- 156 module families (28 spanning 5+, 7 spanning 10+, 1 spanning 21) — verified in NB05 output
- 6,691 predictions (2,455 family-backed, 4,236 module-only) — verified with per-organism files
- Benchmark (95.8% ortholog, 29.1% domain, <1% module-ICA strict) — confirmed across 32 organisms with standard deviations
Limitations well-acknowledged: The KO-level precision limitation is explicitly discussed with quantitative evidence (DvH: 1.2 genes per unique KO). Module-ICA is characterized as "complementary" to sequence-based methods. The D'Agostino K² failure is documented with before/after metrics.
Areas for improvement:
1. Prediction granularity gap: The README states predictions should be interpreted as "process-level context," but many top predictions are domain-family labels (e.g., "DEAD helicase", "LacI", "Response_reg", "ABC_tran", "Flg_bb_rod") inherited from PFam enrichment. These are closer to molecular function than biological process. The 84% ID-to-description resolution (NB06 cell 6) helps readability but doesn't change the semantic level.
-
No confidence threshold guidance: Predictions span confidence 0.41–14.77 (median 1.32) with no calibration against known annotations or recommended filtering threshold.
-
Low-quality organism modules: Methanococcus_JJ (52.9% enriched, 9/17 modules) and ANA3 (60.5%, 23/38) show markedly lower validation rates. These organisms' predictions may be less reliable, but no quality flag distinguishes them.
Suggestions
-
[High] Add outputs or summaries to NB01–02. These 18 code cells have zero saved outputs. Options: (a) re-run with
nbconvert --execute --inplaceto capture outputs, (b) add markdown summary tables of key results. This would let readers understand the data landscape without JupyterHub access. -
[High] Add confidence threshold guidance. A brief analysis of prediction precision vs. confidence threshold on the held-out set — or even a simple statement like "predictions with confidence > X represent the top quartile" — would help downstream users filter predictions.
-
[Medium] Distinguish domain-level from process-level predictions. Consider post-processing predictions into two tiers: (a) process-level from SEED/KEGG enrichment, and (b) domain-level from PFam/TIGRFam. This would make the output more useful and honest about granularity.
-
[Medium] Apply FDR correction to cofitness validation. Apply BH correction (already used in NB04) to the 1,114 Mann-Whitney p-values for methodological consistency. Given the strong effect sizes (mean 3.0× enrichment ratio), the result should be robust.
-
[Low] Sort ortholog BBH hits by ratio before selection. In
run_benchmark.py, add.sort_values('ratio', ascending=False)to the ortholog lookup to ensure the highest-quality ortholog is used. -
[Low] Flag organisms with weak module quality. Consider adding a quality flag for organisms below a minimum enrichment threshold (e.g., <70% modules enriched) so downstream users can weight predictions accordingly.
-
[Low] Rename
pilot_organisms.csvtoselected_organisms.csv. The file contains all 32 selected organisms, not the initial "~5 pilots" described in NB01. The variable naming (pilot_idsin NB03–06 vs.org_idsin NB07) creates a minor inconsistency. -
[Nice-to-have] Include a summary table of largest module families. The README mentions families spanning 10+ and 21 organisms but doesn't name them. A supplementary table of the top families with consensus annotations would be valuable for biologists.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Benchmark Neighborhood
Benchmark Strict
Enrichment Summary
Module Families
Module Size Distribution
Pca Eigenvalues
Prediction Summary
Validation Summary
Notebooks
01_explore_and_select.ipynb
01 Explore And Select
View notebook →
02_extract_matrices.ipynb
02 Extract Matrices
View notebook →
03_ica_modules.ipynb
03 Ica Modules
View notebook →
04_module_annotation.ipynb
04 Module Annotation
View notebook →
05_cross_organism_alignment.ipynb
05 Cross Organism Alignment
View notebook →
06_function_prediction.ipynb
06 Function Prediction
View notebook →
07_benchmarking.ipynb
07 Benchmarking
View notebook →