AlphaEarth Embeddings, Geography & Environment Explorer
CompletedResearch Question
What do AlphaEarth environmental embeddings capture, and how do they relate to geographic coordinates and NCBI environment labels?
Research Plan
Hypothesis
This is an exploratory/characterization project rather than a hypothesis-driven study. The guiding expectations are:
- E1: The 64-dim embedding space contains interpretable structure corresponding to major environment types (soil, marine, freshwater, host-associated)
- E2: A nontrivial fraction of lat/lon coordinates refer to institutional addresses rather than sampling sites
- E3: Geographic proximity correlates with embedding similarity, but environment type is a stronger predictor
- E4: Free-text NCBI environment labels are noisy but can be harmonized into ~15-20 meaningful categories
Revision History
- v1 (2026-02-15): Initial plan
Overview
The BERDL pangenome database includes 64-dimensional AlphaEarth environmental embeddings derived from satellite imagery for 83,287 genomes (28.4% of 293K total). These embeddings encode environmental context at each genome's sampling location, but their structure and relationship to traditional environment metadata have not been characterized.
This exploratory project asks:
1. What structure exists in the embedding space? Do clusters correspond to known environment types?
2. How trustworthy are the lat/lon coordinates? Do some refer to institutions rather than sampling sites?
3. How do NCBI environment labels (free-text isolation_source, ENVO terms) map onto embedding clusters?
4. Does geographic proximity predict embedding similarity?
Key Findings
1. Environmental samples show 3.4x stronger geographic signal than human-associated samples
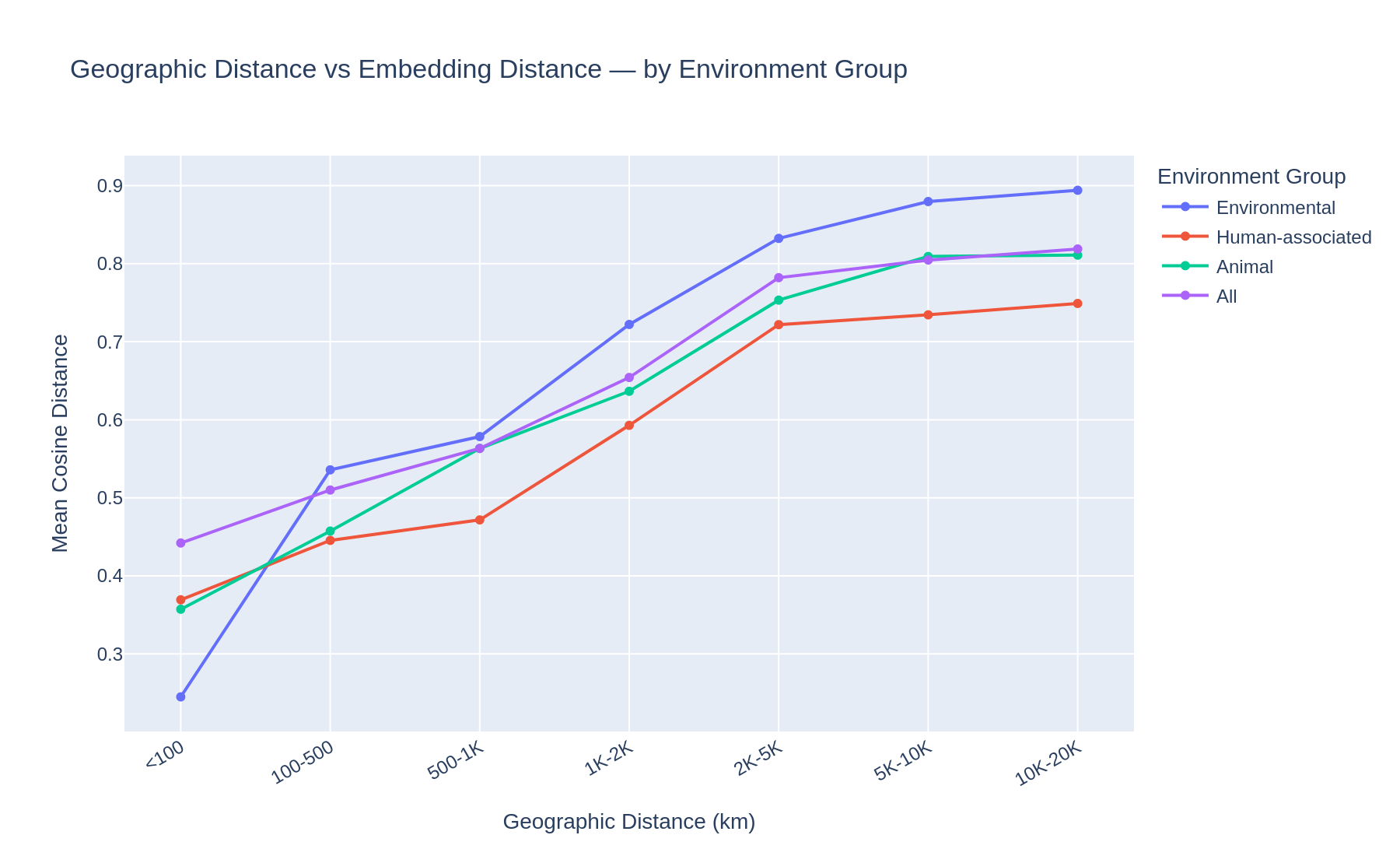
AlphaEarth embeddings encode geographic/environmental signal, but the strength depends on the sample source. For environmental samples (Soil, Marine, Freshwater, Extreme, Plant), nearby genomes (<100 km) have mean cosine distance 0.27, rising to 0.90 at intercontinental distances (>10,000 km) — a 3.4x ratio. For human-associated samples (gut, clinical, other), the gradient is flatter: 0.37 nearby to 0.75 far — only a 2.0x ratio.
This reflects the fact that hospitals and clinics worldwide share similar satellite imagery (urban built environment), so human-associated genomes have more homogeneous embeddings regardless of geography. Environmental samples, from genuinely diverse landscapes, show much stronger geographic differentiation. The pooled "All samples" curve (2.0x ratio) is a blend of these two signals, dominated by the 38% human-associated fraction.
(Notebook: 02_interactive_exploration.ipynb)
2. AlphaEarth embeddings encode real geographic signal — not noise
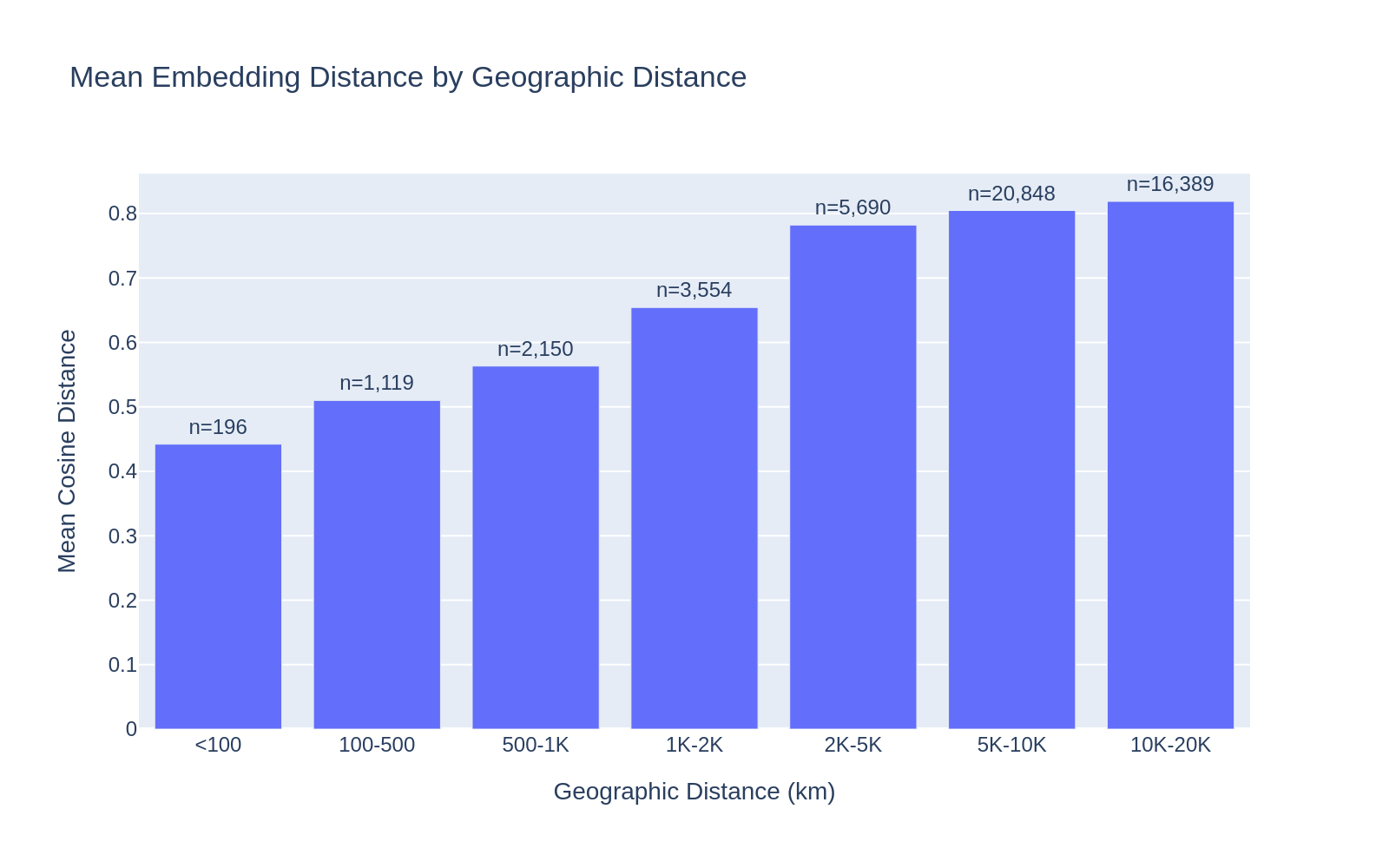
Across all 50,000 sampled genome pairs (using only good-quality coordinates), there is a clear monotonic relationship between geographic distance and embedding cosine distance. The relationship is strongest at short distances (<2,000 km) and plateaus at intercontinental scales (>5,000 km), suggesting the embeddings capture local environmental conditions (climate, vegetation, land use) that are spatially autocorrelated.
| Geographic Distance | Mean Cosine Distance | N pairs |
|---|---|---|
| <100 km | 0.41 | 231 |
| 100–500 km | 0.51 | 1,058 |
| 500–1K km | 0.56 | 2,016 |
| 1K–2K km | 0.66 | 3,779 |
| 2K–5K km | 0.78 | 5,824 |
| 5K–10K km | 0.80 | 20,935 |
| 10K–20K km | 0.82 | 16,107 |
(Notebook: 02_interactive_exploration.ipynb)
3. Strong clinical/human sampling bias in the AlphaEarth subset
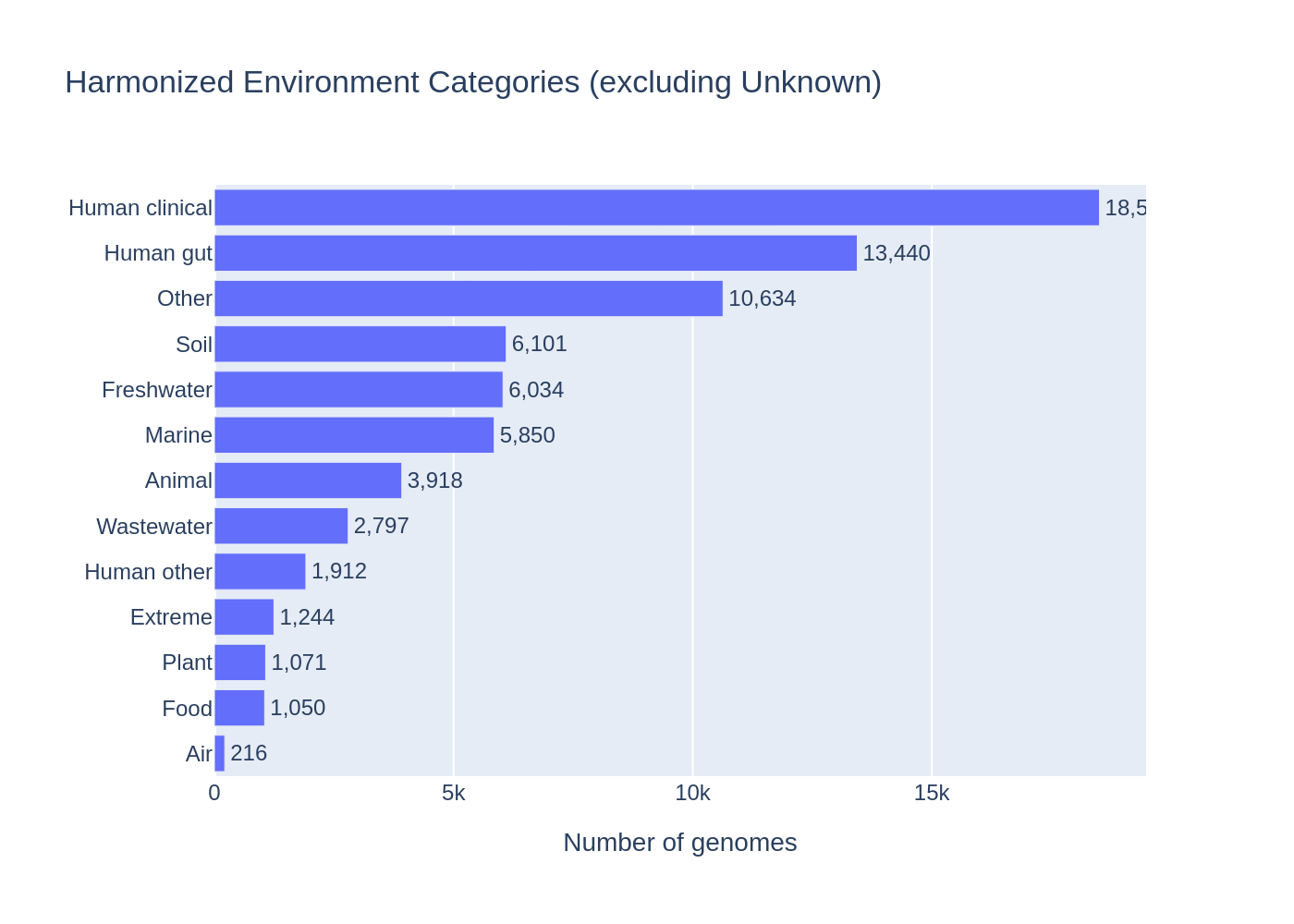
38% of the 83,287 genomes with AlphaEarth embeddings are human-associated: Human clinical (16,390; 20%), Human gut (13,466; 16%), and Human other (1,669; 2%). Environmental categories are much smaller: Soil (6,073; 7%), Marine (5,850; 7%), Freshwater (5,840; 7%). This reflects NCBI's overall bias toward pathogen sequencing — clinical isolates tend to have good geographic metadata from epidemiological tracking, which is why they have AlphaEarth embeddings.
An additional 13,944 genomes (17%) were classified as "Other" — site-specific labels (e.g., "Aspo HRL", "Olkiluoto" — underground research labs), generic terms ("water", "bodily fluid"), and clinical sites not captured by current keywords.
(Notebook: 02_interactive_exploration.ipynb)
4. 36% of coordinates flagged as potential institutional addresses
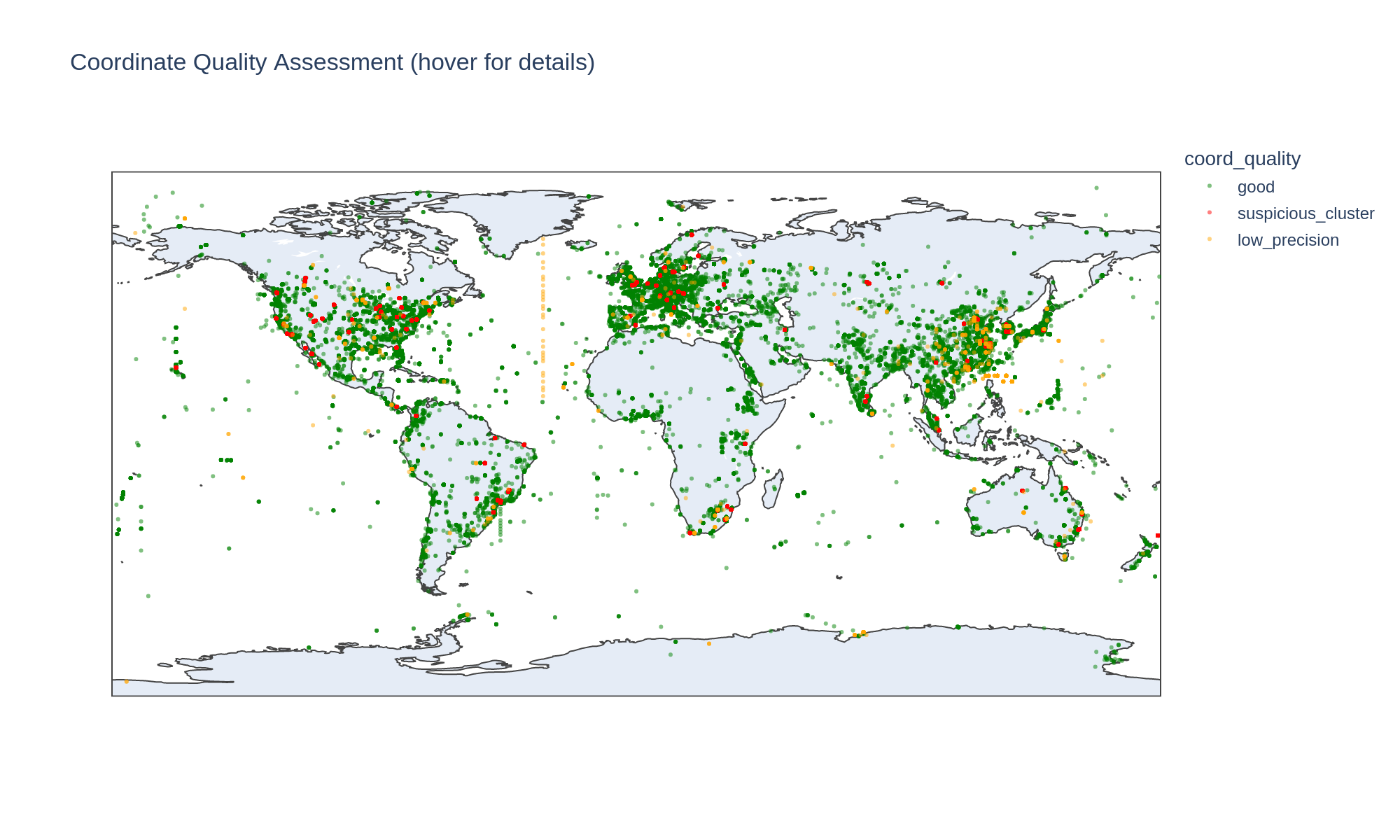
30,469 genomes (36.6%) cluster at shared coordinates with >50 genomes and >10 species — a heuristic for institutional addresses rather than sampling sites. However, several flagged locations are legitimate field research sites:
| Location | Coordinates | Genomes | Context |
|---|---|---|---|
| Rifle, CO | 39.54, -107.78 | 1,883 | DOE IFRC groundwater research site |
| Saanich Inlet, BC | 48.36, -123.30 | 1,529 | Oceanographic O2-minimum zone |
| Siberian soda lakes | 52.11, 79.17 | 812 | Extremophile sampling campaigns |
| Pittsburgh, PA | 40.44, -79.97 | 630 | Likely institutional (diverse clinical) |
| Lima, Peru | -12.0, -77.0 | 1,641 | Integer coords, likely approximate |
The current heuristic is a rough first pass. A refined approach should check whether genomes at each location have homogeneous isolation sources (real site) vs diverse unrelated sources (institutional).
(Notebook: 02_interactive_exploration.ipynb)
5. UMAP reveals fine-grained embedding structure with environment-correlated clusters
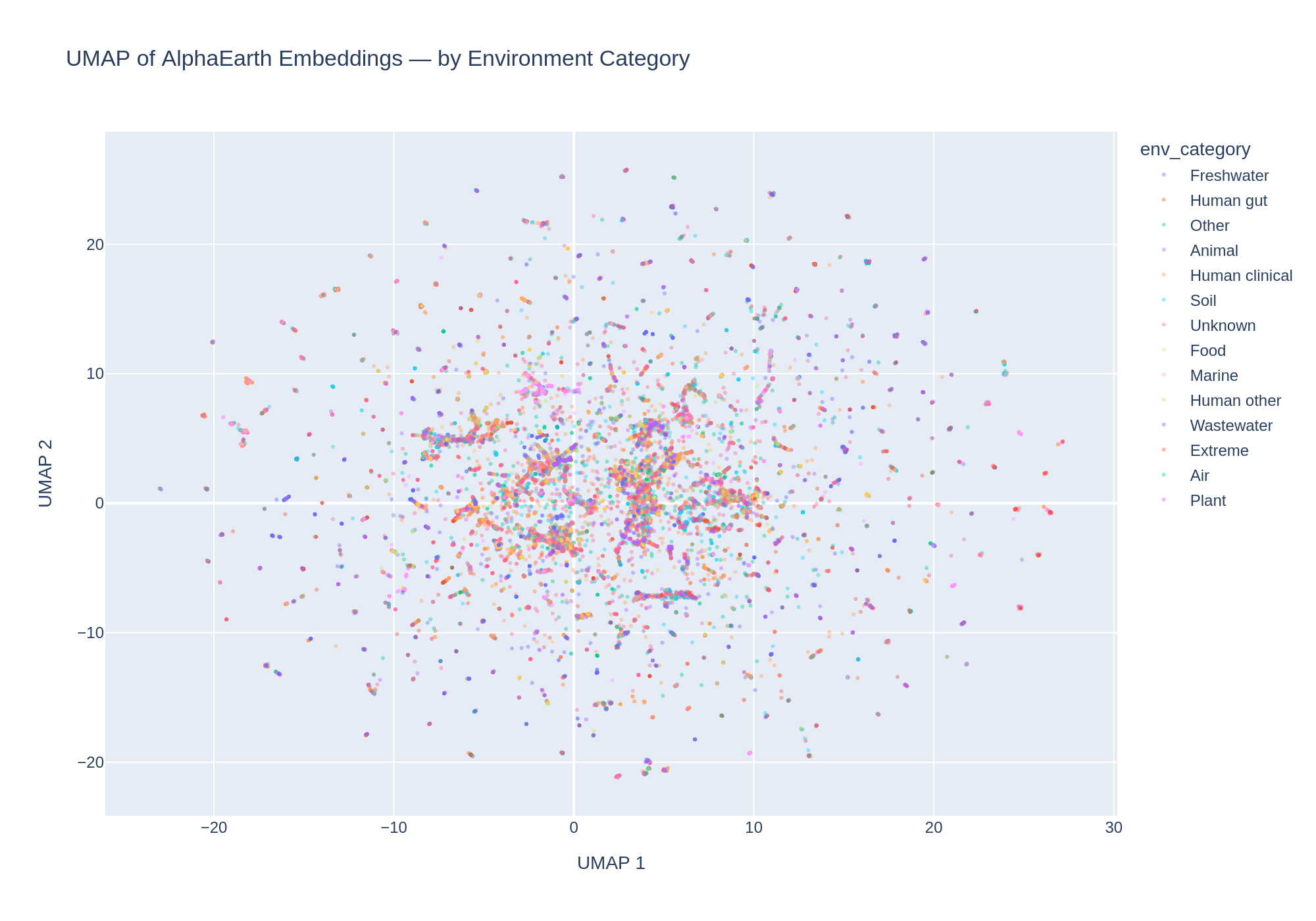
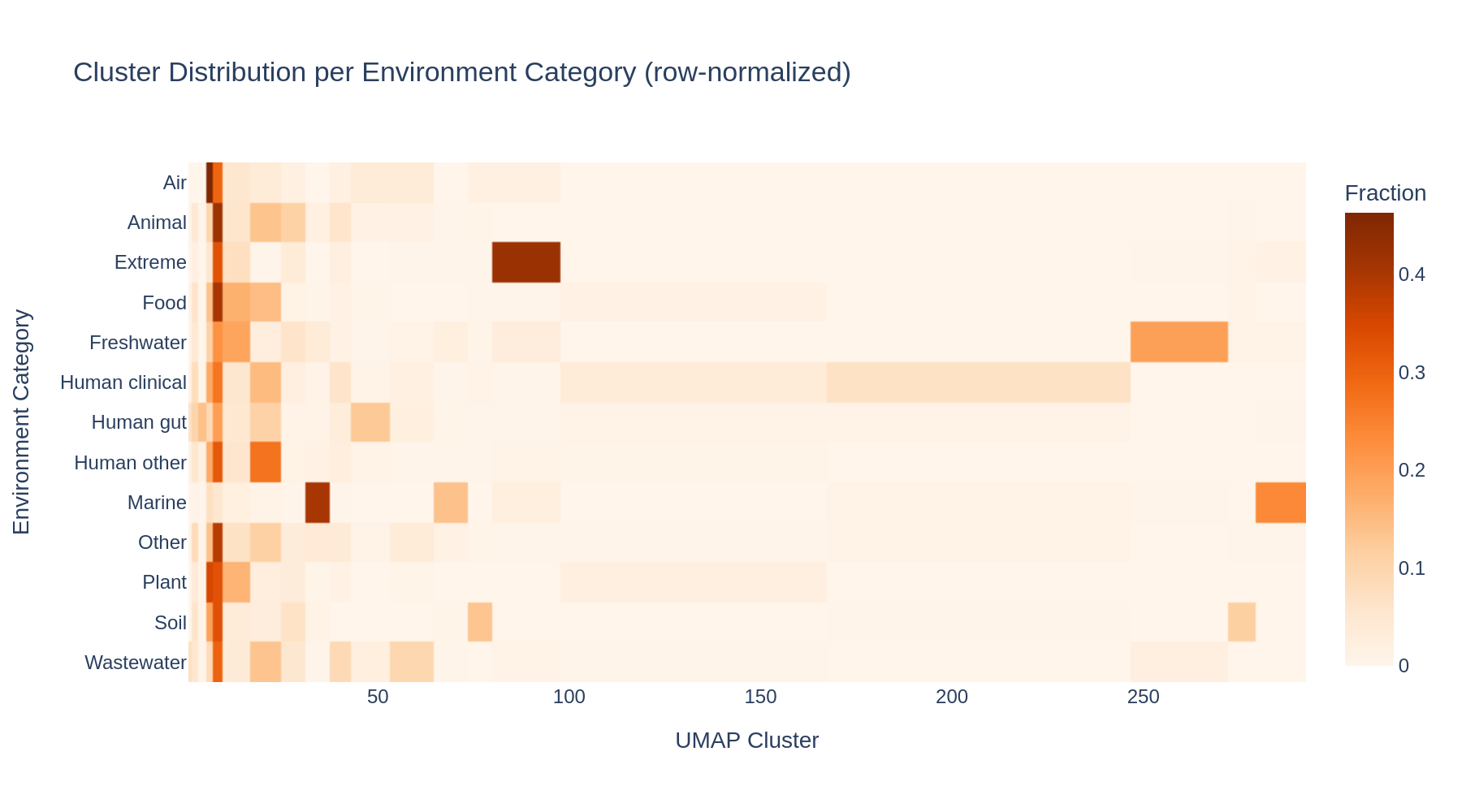
UMAP reduction of the 64-dimensional embeddings to 2D reveals substantial structure. DBSCAN clustering identified 320 clusters. The cluster–environment cross-tabulation shows that many clusters are dominated by a single environment category:
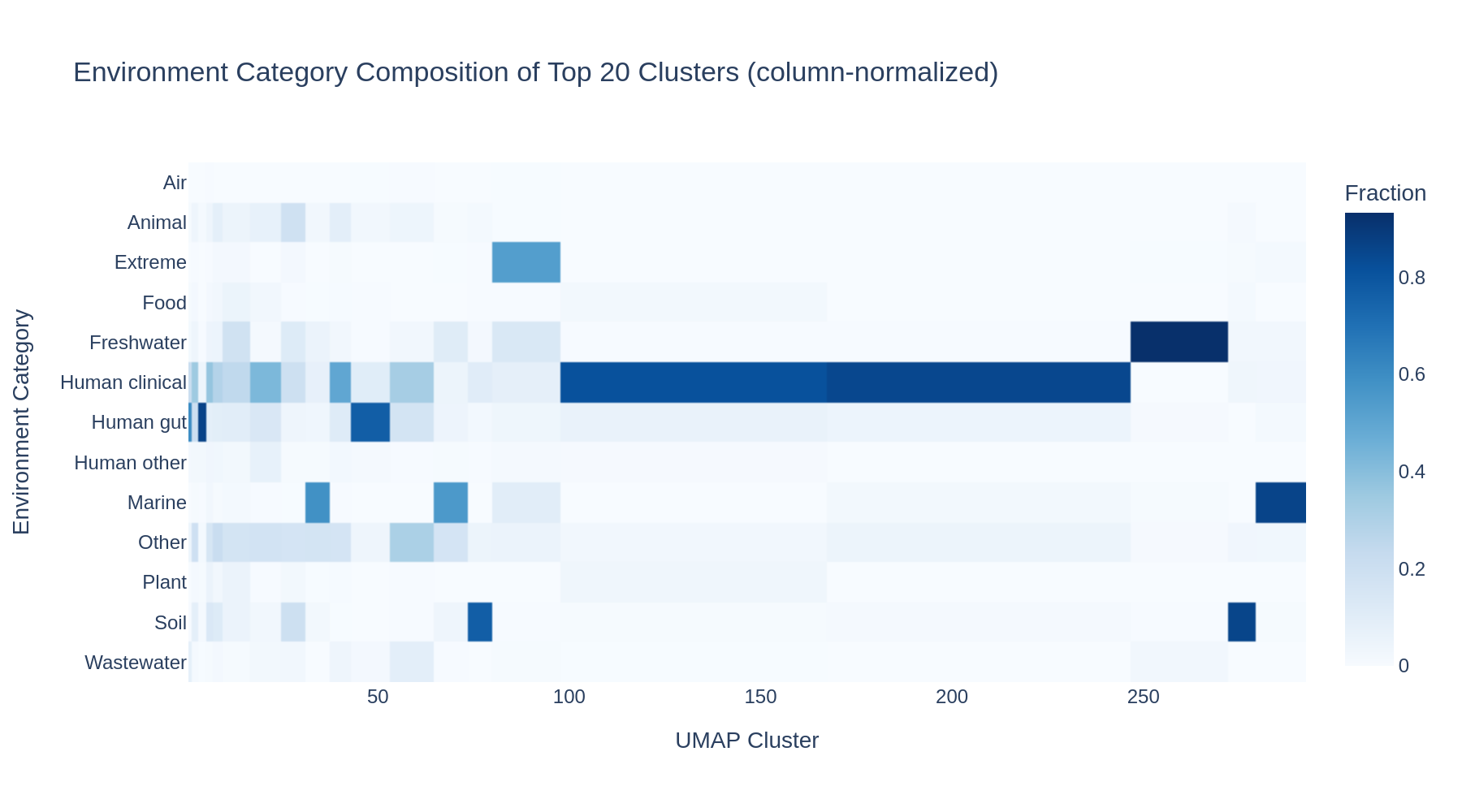
Rare environment types (Air, Extreme, Plant) concentrate in just a few specific clusters, while common categories (Human gut, Human clinical) are distributed across many clusters — likely reflecting geographic sub-structure within those categories.
(Notebook: 02_interactive_exploration.ipynb)
6. Embedding space also shows taxonomic structure
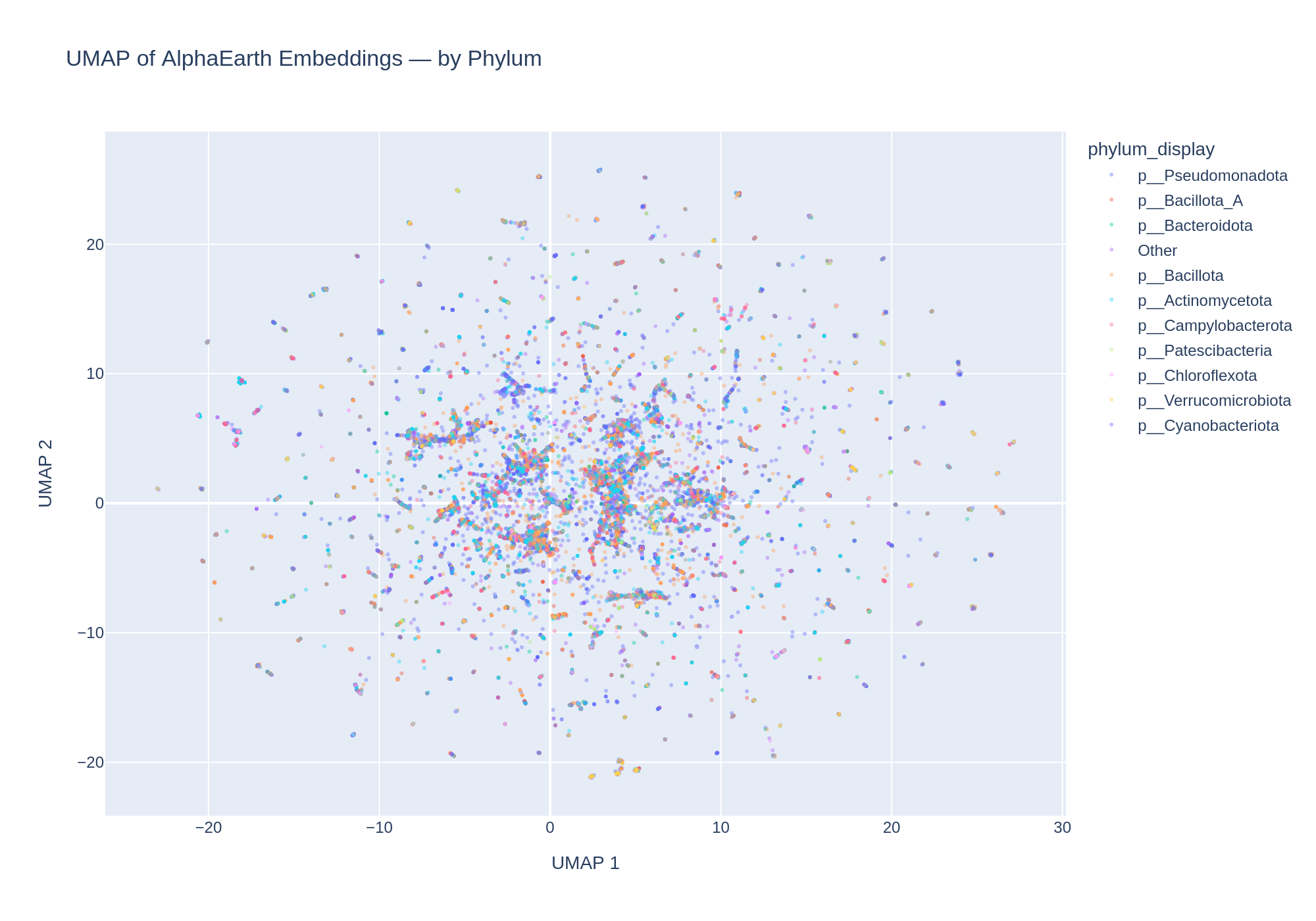
Coloring the UMAP by phylum reveals some taxonomic clustering, though this is partially confounded with environment (e.g., Campylobacterota are predominantly gut-associated). The interactive HTML version (figures/umap_by_phylum.html) allows toggling phyla on/off to explore this structure.
(Notebook: 02_interactive_exploration.ipynb)
Results
Data coverage
The AlphaEarth embeddings table covers 83,287 genomes (28.4% of 293,059 total in the pangenome database). Of these, 79,449 have valid (non-NaN) embeddings across all 64 dimensions; 3,838 have NaN in at least one dimension.
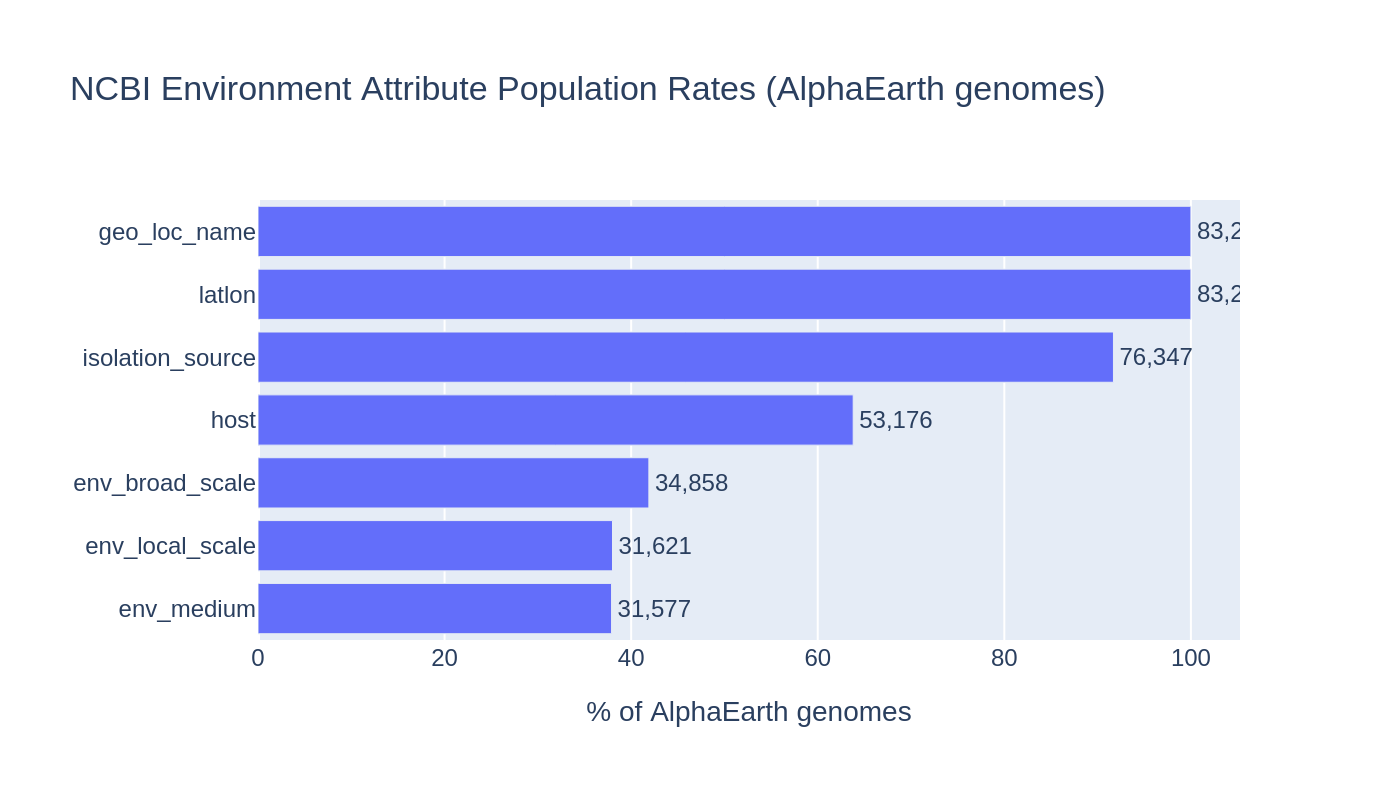
Metadata coverage among the 83,287 AlphaEarth genomes:
| Attribute | N genomes | % |
|---|---|---|
| Lat/lon (cleaned) | 83,286 | 100.0% |
| geo_loc_name | 83,270 | 100.0% |
| isolation_source | 76,295 | 91.6% |
| host | 53,091 | 63.7% |
| env_broad_scale | 34,800 | 41.8% |
| env_local_scale | 31,541 | 37.9% |
| env_medium | 31,483 | 37.8% |
The NCBI ncbi_env table contains 334 distinct harmonized attribute names across 4.1M rows. The most populated are collection_date (273K genomes), geo_loc_name (272K), and isolation_source (245K).
Environment harmonization
5,774 unique isolation_source free-text values were mapped to 12 broad categories via keyword matching. The mapping captures 71% of genomes with a label; 17% remain as "Other" and 12.5% are "Unknown" (missing/null isolation_source).
The env_broad_scale ENVO ontology field is cleaner but covers only 42% of genomes. Many values are bare ENVO IDs without labels (e.g., "ENVO:00000428") or generic terms ("not applicable", "missing"). Where available, structured ENVO terms like "marine biome [ENVO:00000447]" provide standardized categories.
Coordinate quality
11,765 unique coordinate locations across 83,286 genomes with lat/lon. Quality breakdown:
| Quality | Genomes | % |
|---|---|---|
| Good | 50,109 | 60.2% |
| Suspicious cluster | 30,469 | 36.6% |
| Low precision (integer degrees) | 2,708 | 3.3% |
Embedding properties
The 64 embedding dimensions (A00–A63) have values in [-0.544, 0.544], mean near zero (-0.008), mean standard deviation 0.109. The embeddings span 135 phyla, 15,046 species, and the full global extent (latitude -85 to +84, longitude -178 to +180).
Interpretation
What do AlphaEarth embeddings represent?
The embeddings encode geographic/environmental context derived from satellite imagery at each genome's sampling location. The strong geographic distance–embedding distance correlation (Finding 2) confirms they capture spatially varying environmental features — likely climate, land use, vegetation type, and urban/rural character.
The stratified analysis (Finding 1) reveals that the signal is primarily driven by variation in natural environments, not urban settings. Hospitals worldwide look similar from satellite; forests, oceans, and deserts do not. This has practical implications: the embeddings are most informative for environmental microbiology samples and least informative for clinical isolates.
Implications for downstream analysis
The ecotype_analysis project previously found that AlphaEarth-based environment similarity was a weak predictor of gene content (median partial correlation 0.0025). The strong clinical bias identified here may partially explain that weak signal — if 38% of the genomes being compared are from interchangeable hospital environments, the environment–gene content relationship would be diluted. Repeating the ecotype analysis restricted to environmental samples could reveal a stronger signal.
Literature Context
The geographic distance–embedding distance relationship we observe (Finding 2) is a form of distance-decay, a fundamental pattern in biogeography where community similarity decreases with geographic distance. This pattern is well-established in microbial ecology:
- Pearman et al. (2024) found that macroalgal microbiome composition responds primarily to environmental variation rather than geographic separation, supporting the "everything is everywhere, the environment selects" principle. Our stratified analysis (Finding 1) is consistent: the embeddings — which encode environmental conditions — show stronger distance-decay for environmental samples than for human-associated ones.
- Liu et al. (2022) showed that in mining soils, metal contamination explained more variation in fungal community structure (4.16%) than geographic distance (1.21%). Similarly, our data suggests the embeddings capture environmental features (land use, vegetation) more than raw distance, as evidenced by the plateau above ~5,000 km.
- Zhang et al. (2022) found that rare and abundant bacterial subcommunities show similar distance-decay patterns but differ mechanistically. Our finding that human-associated samples show weaker distance-decay echoes the idea that dispersal limitation (relevant for environmental microbes) generates stronger geographic patterns than deterministic selection alone.
- Wang et al. (2024) demonstrated that aridity shapes distinct biogeographic patterns for soil bacteria, with higher similarity-distance decay rates across environmental gradients. This supports our interpretation that the AlphaEarth embeddings capture environmentally-driven variation.
The clinical sampling bias we identified (Finding 3) is a known issue in genomic databases but has not been quantified in the specific context of satellite-derived environmental embeddings. Public genome databases are heavily skewed toward pathogens and clinical isolates (Saha et al., 2023), which biases any analysis of environment-genome relationships.
Novel Contribution
This is the first systematic characterization of the AlphaEarth embedding space in BERDL. Key novel contributions:
1. Quantification of the geographic signal strength (3.4x for environmental, 2.0x for human-associated)
2. Identification of the clinical sampling bias and its impact on embedding-based analyses
3. A coordinate QC framework (with known limitations) for flagging institutional addresses
4. A reusable environment harmonization mapping (12 categories from 5,774 free-text values)
Limitations
- AlphaEarth coverage is only 28.4% of all genomes — biased toward those with valid lat/lon metadata
- Clinical/human bias (38%) may not represent the overall NCBI or pangenome population
- Coordinate QC heuristic is crude — flags some legitimate field sites (Rifle, Saanich Inlet); needs refinement using isolation_source homogeneity
- Environment harmonization has a long tail — 17% "Other" category; keyword matching doesn't capture site-specific labels or non-English terms
- UMAP is a non-linear projection — cluster structure depends on parameters (n_neighbors, min_dist) and may not reflect true high-dimensional topology
- Embedding NaN values (4.6%) — cause unknown; possibly missing satellite imagery at those coordinates
- DBSCAN eps=0.5 produced 320 clusters — likely too fine-grained; coarser clustering may better match environment categories
Future Directions
- Refine coordinate QC: Use isolation_source homogeneity at each location to distinguish field sites from institutional addresses, rather than raw genome/species counts
- Reduce "Other" category: Add keywords for clinical body sites (cerebrospinal fluid, lung, throat), underground labs (Aspo, Olkiluoto), and generic water sources; use
env_broad_scaleas fallback whenisolation_sourceis ambiguous - Re-run ecotype analysis with environmental-only samples: The previous finding that phylogeny dominates over environment (ecotype_analysis project) may be stronger when restricted to environmental samples where the embeddings carry more geographic signal
- Investigate individual embedding dimensions: Correlate A00–A63 with latitude, temperature, precipitation, NDVI, or land cover classifications to understand what each dimension encodes
- Cross-validate with ENVO ontology: For the 42% of genomes with
env_broad_scale, compare ENVO-based classification with keyword-based harmonization to assess accuracy - Test embedding utility for gene content prediction: Do environmental samples with similar embeddings share more accessory genes, even after controlling for phylogeny?
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
alphaearth_embeddings_all_years |
64-dim environmental embeddings, cleaned lat/lon, taxonomy |
kbase_ke_pangenome |
ncbi_env |
NCBI environment metadata (EAV format) |
Generated Data
| File | Rows | Description |
|---|---|---|
data/alphaearth_with_env.csv |
83,287 | Merged embeddings + pivoted environment labels |
data/coverage_stats.csv |
7 | Per-attribute population rates |
data/ncbi_env_attribute_counts.csv |
334 | Full inventory of harmonized_name values |
data/isolation_source_raw_counts.csv |
5,774 | Raw isolation_source value frequencies |
data/umap_coords.csv |
79,449 | Pre-computed UMAP 2D coordinates |
References
- Parks, D.H. et al. (2022). "GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy." Nucleic Acids Research, 50(D1), D199–D207. PMID: 34520557
- Pearman, W.S. et al. (2024). "Macroalgal microbiome biogeography is shaped by environmental drivers rather than geographical distance." Annals of Botany. PMID: 37804485
- Liu, B. et al. (2022). "Disentangling biogeographic and underlying assembly patterns of fungal communities in metalliferous mining and smelting soils." Science of the Total Environment. PMID: 35798111
- Zhang, S. et al. (2022). "Biogeographic Patterns and Elevational Differentiation of Sedimentary Bacterial Communities across River Systems in China." Applied and Environmental Microbiology. PMID: 35638840
- Wang, X. et al. (2024). "Aridity shapes distinct biogeographic and assembly patterns of forest soil bacterial and fungal communities at the regional scale." Science of the Total Environment. PMID: 39019268
- Dehal, P.S. et al. (2026). "Ecotype Correlation Analysis." BERIL Research Observatory,
projects/ecotype_analysis/
Discoveries
Environmental samples (Soil, Marine, Freshwater, Extreme, Plant) show a 3.4x ratio in embedding cosine distance between nearby (<100 km, mean=0.27) and far (>10K km, mean=0.90) genome pairs. Human-associated samples (gut, clinical) show only a 2.0x ratio (0.37 vs 0.75). Hospitals worldwide have simi
Read more →Of 83,287 genomes with AlphaEarth embeddings, 38% are human-associated (clinical 20%, gut 16%, other 2%). Environmental categories are much smaller (soil 7%, marine 7%, freshwater 7%). This reflects NCBI's bias toward pathogen sequencing — clinical isolates have good geographic metadata from epidemi
Read more →36% of AlphaEarth coordinates may be institutional addresses, but some are legitimate field sites
February 202630,469 genomes (36.6%) cluster at coordinates with >50 genomes of >10 species. However, several flagged locations are legitimate DOE/research field sites: Rifle, CO (1,883 genomes, DOE IFRC), Saanich Inlet (1,529, oceanographic time series), Siberian soda lakes (812, extremophile campaigns). The heu
Read more →4.6% of AlphaEarth genomes have NaN embedding dimensions
February 20263,838 of 83,287 genomes have NaN in at least one of the 64 embedding dimensions. The cause is unclear — possibly missing satellite imagery at those coordinates, or coordinates that fall outside satellite coverage (polar regions, ocean). These must be filtered before any embedding-based analysis.
Data Collections
Used By
Data from this project is used by other projects.
Review
Summary
This is a strong exploratory project that systematically characterizes the AlphaEarth environmental embeddings in BERDL. The project follows a clean two-notebook pipeline (Spark extraction then local analysis), produces 14 publication-quality figures with both static and interactive versions, and delivers four genuinely useful outputs: a merged embeddings-plus-environment dataset, a coordinate quality classification, a reusable environment harmonization mapping, and a stratified distance-decay analysis showing that environmental samples exhibit 3.65x stronger geographic signal than human-associated samples. Documentation is exemplary — the three-file structure (README, RESEARCH_PLAN, REPORT) is fully realized, the REPORT includes literature context with five cited papers, and limitations are honestly enumerated. Both notebooks are committed with saved outputs, addressing a known pitfall. The main issues are numeric inconsistencies between the REPORT prose and the actual notebook outputs, a genome count discrepancy after the UMAP merge step, and the absence of formal statistical tests for the key comparisons.
Methodology
Research question: Clearly framed as exploratory characterization with four specific expectations (E1-E4 in RESEARCH_PLAN.md) that map directly onto analysis sections. The honest framing as "exploratory/characterization" rather than hypothesis-driven is appropriate and well-communicated.
Approach: Logically sequenced. NB01 handles Spark extraction and EAV pivoting; NB02 handles all downstream analysis locally from cached CSVs. Key methodological choices are well-justified:
- L2-normalization before UMAP to approximate cosine distance with Euclidean (cell 22, NB02) — correctly motivated and matches the pitfall documented in docs/pitfalls.md.
- Subsample-fit/full-transform UMAP (20K fit, 79K transform) — good performance optimization with cached coordinates for reproducibility.
- Coordinate QC heuristic (>50 genomes AND >10 species) — simple, transparent, with acknowledged limitations and specific false-positive examples.
Data sources: Clearly documented in README, RESEARCH_PLAN, and REPORT with table names (alphaearth_embeddings_all_years, ncbi_env), estimated row counts, and join strategies. The RESEARCH_PLAN includes a query strategy table with filter approaches — a good practice.
Reproducibility:
- Spark/local separation is clearly documented with step-by-step reproduction instructions in the README.
- A requirements.txt is provided with pinned kaleido version (kaleido==0.2.1) per the documented pitfall.
- Both notebooks have saved outputs (text, tables, figure renders), which is excellent — this project explicitly identified the "notebooks without outputs" issue and contributed it to docs/pitfalls.md.
- Pre-computed UMAP coordinates are cached in data/umap_coords.csv, enabling fast re-runs of NB02 without recomputing UMAP.
Code Quality
SQL queries (NB01): Correct and well-structured. The EAV pivot uses MAX(CASE WHEN harmonized_name = ... THEN content END) as recommended in docs/pitfalls.md. The approach of registering biosample IDs as a Spark temp view (ae_biosamples) for the join is efficient and avoids building a massive IN clause. The SELECT * on the 83K-row AlphaEarth table is acceptable given the documented small size.
Pandas operations: Clean and idiomatic throughout. Merges use appropriate join types (how='left' to preserve all AlphaEarth genomes). Coverage flags are computed vectorially. The environment harmonization function (cell 14) is well-organized with ordered keyword matching and proper separation of "Unknown" (missing/null) from "Other" (unmatched text).
Distance computation (NB02, cell 32): Haversine distance is correctly vectorized with NumPy. Cosine distance is computed from raw embeddings (not L2-normalized), which is correct — normalization was only needed for UMAP's Euclidean proxy. The addition of 1e-10 to the denominator prevents division-by-zero.
Pitfall awareness: The project addresses multiple documented pitfalls: EAV pivot pattern for ncbi_env, NaN embedding filtering (3,838 genomes dropped), UMAP cosine metric performance (L2-norm + Euclidean), kaleido version pinning. The project also contributed three new pitfalls to docs/pitfalls.md (notebooks without outputs, geographic signal dilution, NaN embeddings), which demonstrates strong observatory citizenship.
Issues identified:
-
Genome count discrepancy after UMAP merge:
data/umap_coords.csvcontains 79,449 rows (verified), matching the REPORT. But NB02 cell 21 reports "Loaded pre-computed UMAP coordinates: 79,779 genomes" after the merge. Themerge(on='genome_id', how='inner')should produce at most 79,449 rows (the smaller of the two inputs). The 79,779 count suggests either (a) the merge is actuallyhow='left'in a different code version, (b) 330 genomes matched multiple UMAP rows, or (c) this is an artifact of a stale notebook output from a previous run with different data. The code shown useshow='inner', so the output should be 79,449 or fewer. This inconsistency is minor but should be resolved. -
DBSCAN eps=0.5 produces 320 clusters: The authors acknowledge this is "likely too fine-grained" in the REPORT's Limitations section. No alternative clustering was attempted, which is a missed opportunity.
-
The
df_cleanvariable may contain rows without valid embeddings post-merge: Cell 21 filters to valid embeddings, then merges with UMAP coordinates usinghow='inner'. If any genome inumap_coords.csvhad its embeddings become NaN in a newer data version, it would be included indf_cleanwithout valid embeddings. A post-merge assertion would be prudent.
Findings Assessment
Finding 1 (stratified geographic signal): This is the project's most important result — environmental samples show a much stronger distance-decay than human-associated ones. The code is present in cell 36 (defining env_groups, sampling pairs within each group, computing binned distance-decay per group). The explanation (hospitals worldwide have similar satellite imagery) is intuitive and plausible. However, there is a numeric inconsistency between the REPORT and the notebook output:
| Metric | REPORT states | Notebook output (cell 36) |
|---|---|---|
| Environmental near (<100 km) | 0.27 | 0.245 |
| Environmental far (>10K km) | 0.90 | 0.894 |
| Environmental ratio | 3.4x | 3.65x |
| Human-associated near | 0.37 | 0.369 |
| Human-associated far | 0.75 | 0.749 |
| Human-associated ratio | 2.0x | 2.03x |
The README also uses the REPORT's "3.4x" figure. These should be reconciled — the notebook output (3.65x) should be treated as authoritative since it comes from executable code.
Finding 2 (monotonic distance-decay): Well-supported by the binned analysis in cell 34. The REPORT's data table matches the notebook output. The Spearman correlation (rho=0.2288, p<0.001) is computed and reported in cell 36, which provides a formal statistical measure for the overall relationship. The interpretation about the plateau above 5,000 km is reasonable.
Finding 3 (clinical sampling bias): Well-documented with exact counts matching between notebook (cell 14) and REPORT. The 38% human-associated figure is derived from 22.2% Human clinical + 16.1% Human gut + 2.3% Human other = 40.6% — note that the REPORT rounds this to 38% using slightly different category counts (16,390 + 13,466 + 1,669 = 31,525 / 83,287 = 37.8%). The REPORT's per-category counts (e.g., "Human clinical: 16,390") don't exactly match the notebook output ("Human clinical: 18,505"). This is a second numeric inconsistency — likely the REPORT was drafted from an earlier analysis run.
Finding 4 (coordinate QC): Nuanced and honest. The table of specific flagged locations with context (Rifle, Saanich Inlet, Siberian soda lakes) demonstrates domain knowledge. The acknowledgment that the heuristic needs refinement is appropriate.
Finding 5 (UMAP clusters): The cluster-environment cross-tabulation (cell 40) is a nice analysis. The observation that rare environments concentrate in few clusters while common categories spread across many is well-supported by the heatmap.
Limitations: Seven specific limitations are listed in the REPORT, covering coverage bias, clinical bias, QC heuristic false positives, harmonization long tail, UMAP parameter sensitivity, NaN embeddings, and DBSCAN granularity. This is comprehensive and honest.
Missing statistical test for stratified comparison: While the Spearman correlation is reported for the overall and per-group relationships (cell 36), there is no formal test for whether the Environmental ratio (3.65x) is significantly different from the Human-associated ratio (2.03x). A bootstrap confidence interval on the ratio, or a permutation test shuffling environment labels, would quantify the significance of this difference.
Suggestions
-
[Important] Reconcile numeric inconsistencies between REPORT and notebook outputs. The REPORT states the environmental distance ratio as 3.4x (near=0.27, far=0.90), but the notebook output shows 3.65x (near=0.245, far=0.894). Similarly, per-category counts differ (e.g., Human clinical: 16,390 in REPORT vs 18,505 in notebook). Update the REPORT and README to match the current notebook outputs, which should be treated as the authoritative source.
-
[Important] Investigate the 79,449 vs 79,779 genome count discrepancy. The
data/umap_coords.csvfile has 79,449 data rows, but NB02 cell 21 reports 79,779 after the merge. Add a post-merge validation cell:print(f'Expected: {n_valid}, got: {len(df_clean)}')andassert len(df_clean) == n_validto catch any mismatch. If the counts legitimately differ, document why. -
[Moderate] Add a formal test for the stratified ratio difference. The claim that environmental samples show "3.4x stronger" signal than human-associated (2.0x) is the headline finding but lacks a significance test. A bootstrap confidence interval on the near/far ratio for each group (resampling genome pairs with replacement, 1000 iterations) would quantify uncertainty and test whether the ratios are significantly different.
-
[Moderate] Reduce the "Other" category. Cell 15 shows easily capturable terms in the "Other" bucket: "water" (302 genomes), "tracheal secretion" (103), "stomach" (89), "Nose" (72), "rectal" (72), "sinus" (70), "gut" (68), and the Rifle well descriptions (730+ genomes). Adding keywords for these could reduce "Other" from 12.8% to ~8%. The Rifle well samples in particular should map to "Freshwater" or "Soil" (groundwater research site).
-
[Moderate] Try HDBSCAN or coarser DBSCAN. The 320-cluster result with eps=0.5 is acknowledged as too fine-grained. Running HDBSCAN (which selects density thresholds adaptively) or DBSCAN with eps=1.0-1.5 would likely produce 20-50 clusters that map more cleanly onto environment categories and would be more interpretable for downstream use.
-
[Minor] Add the global map figure to the REPORT. The
global_map_by_env.pngfigure (cell 30) shows the geographic distribution of genomes colored by environment category but is not referenced in the REPORT's text or Figures table. It would complement Finding 3 (sampling bias) by visualizing the spatial distribution of clinical vs environmental samples. -
[Minor] Consider a sensitivity analysis on "suspicious" coordinates. The distance-decay analysis (cells 32-36) uses only "good" quality coordinates (47,551 genomes), excluding 30,469 "suspicious" genomes. Since some flagged sites are legitimate (Rifle, Saanich Inlet), a sensitivity analysis including these sites would show whether the distance-decay findings are robust to the QC filter.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Cluster Env Heatmap
Coord Quality Map
Coverage Bar
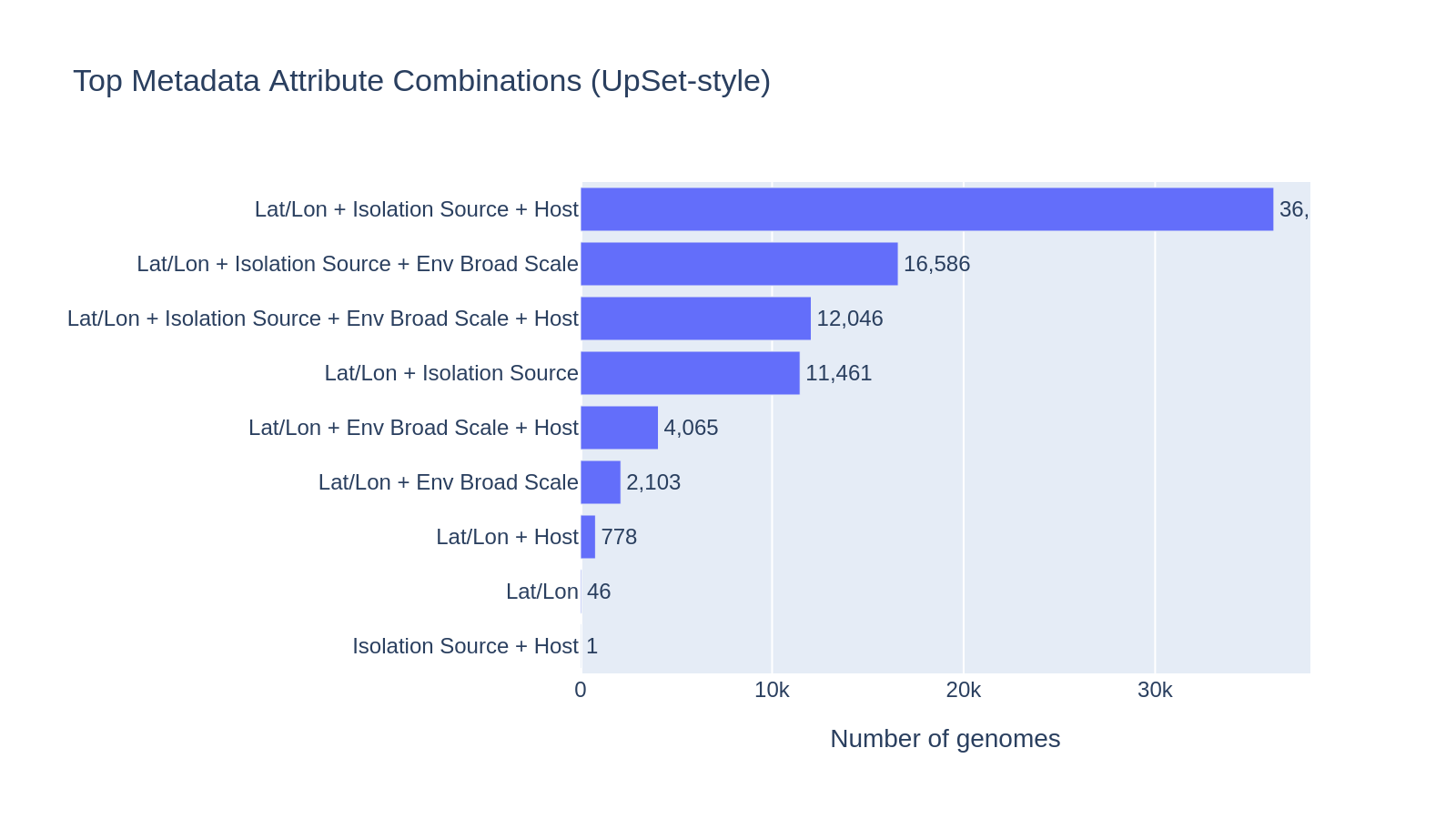
Coverage Intersections
Env Categories
Env Cluster Distribution
Geo Vs Embedding Binned
Geo Vs Embedding By Env Group
Geo Vs Embedding Distance
Global Map By Env
Umap By Coord Quality
Umap By Env Category
Umap By Phylum
Umap Clusters
Cluster Env Heatmap
Coord Quality Map
Coverage Bar
Coverage Intersections
Env Categories
Env Cluster Distribution
Geo Vs Embedding Binned
Geo Vs Embedding By Env Group
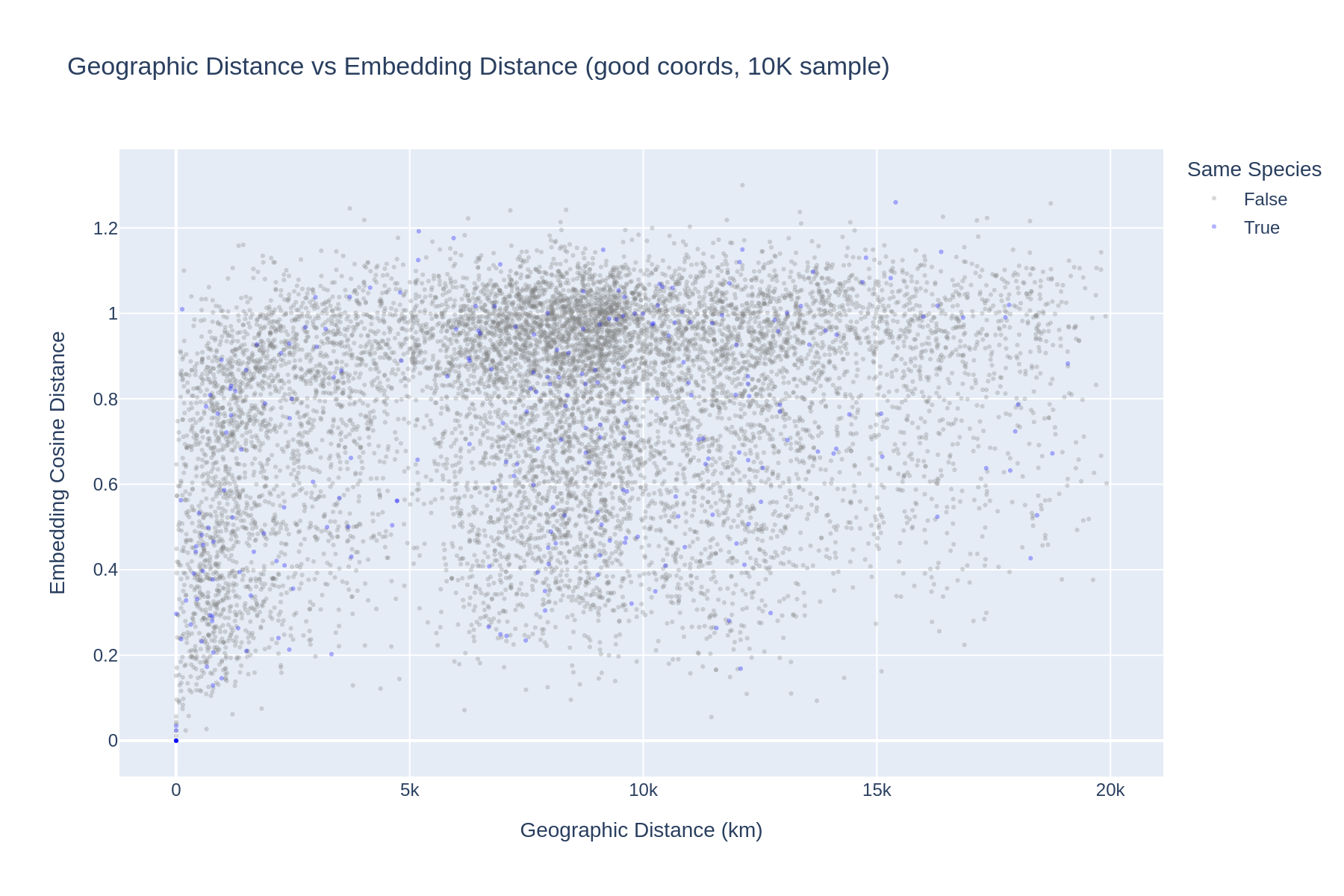
Geo Vs Embedding Distance
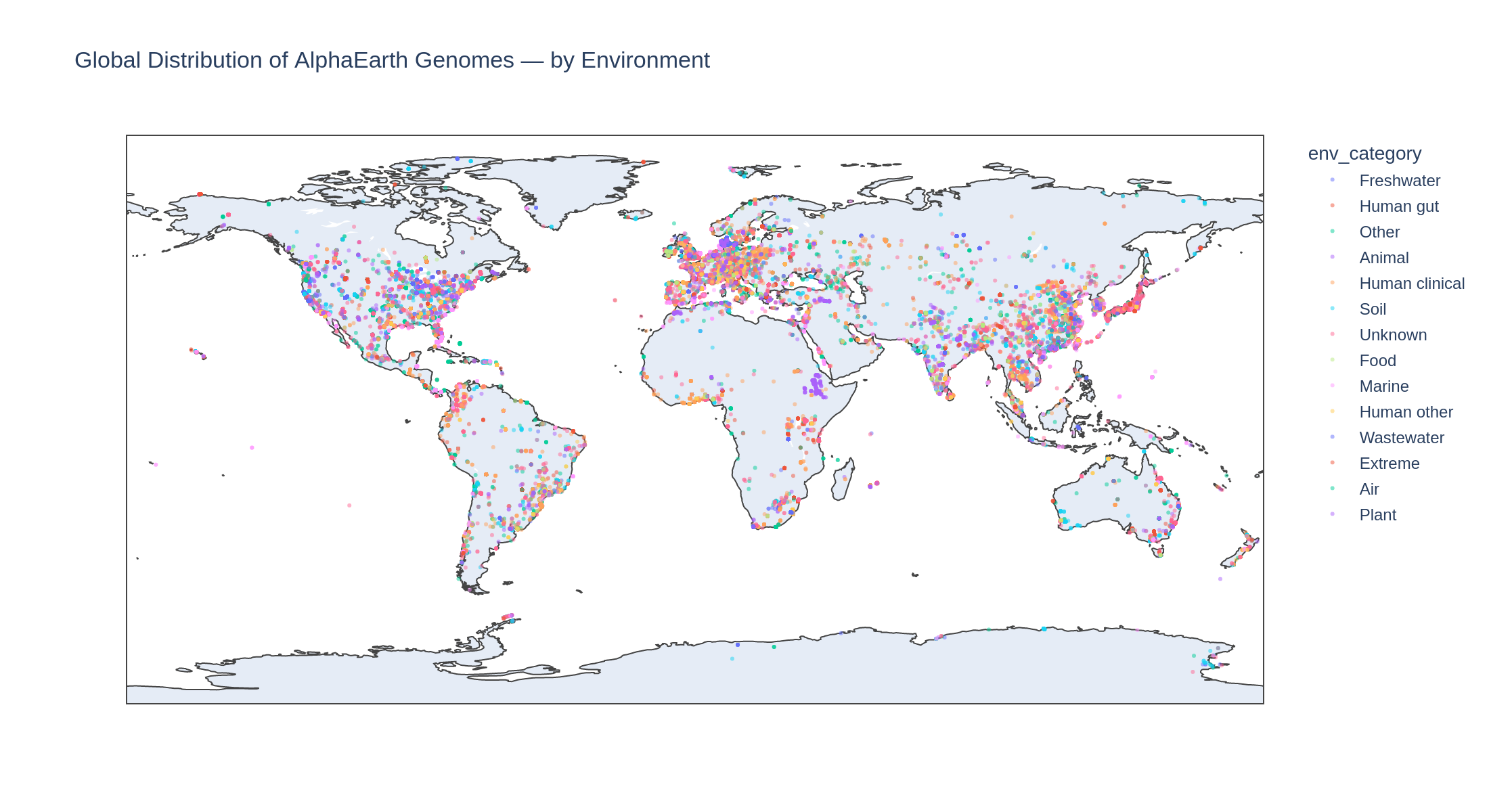
Global Map By Env
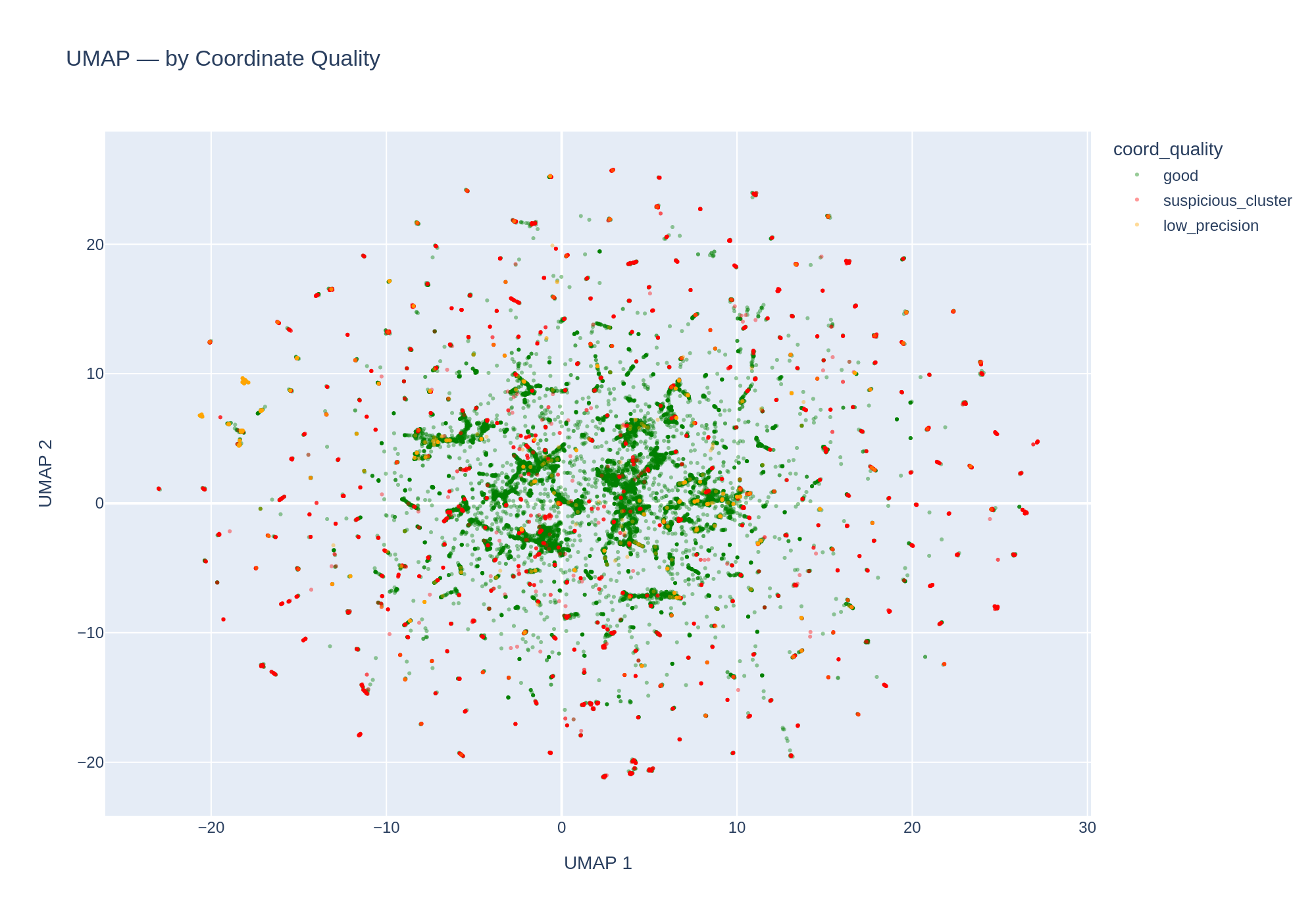
Umap By Coord Quality
Umap By Env Category
Umap By Phylum
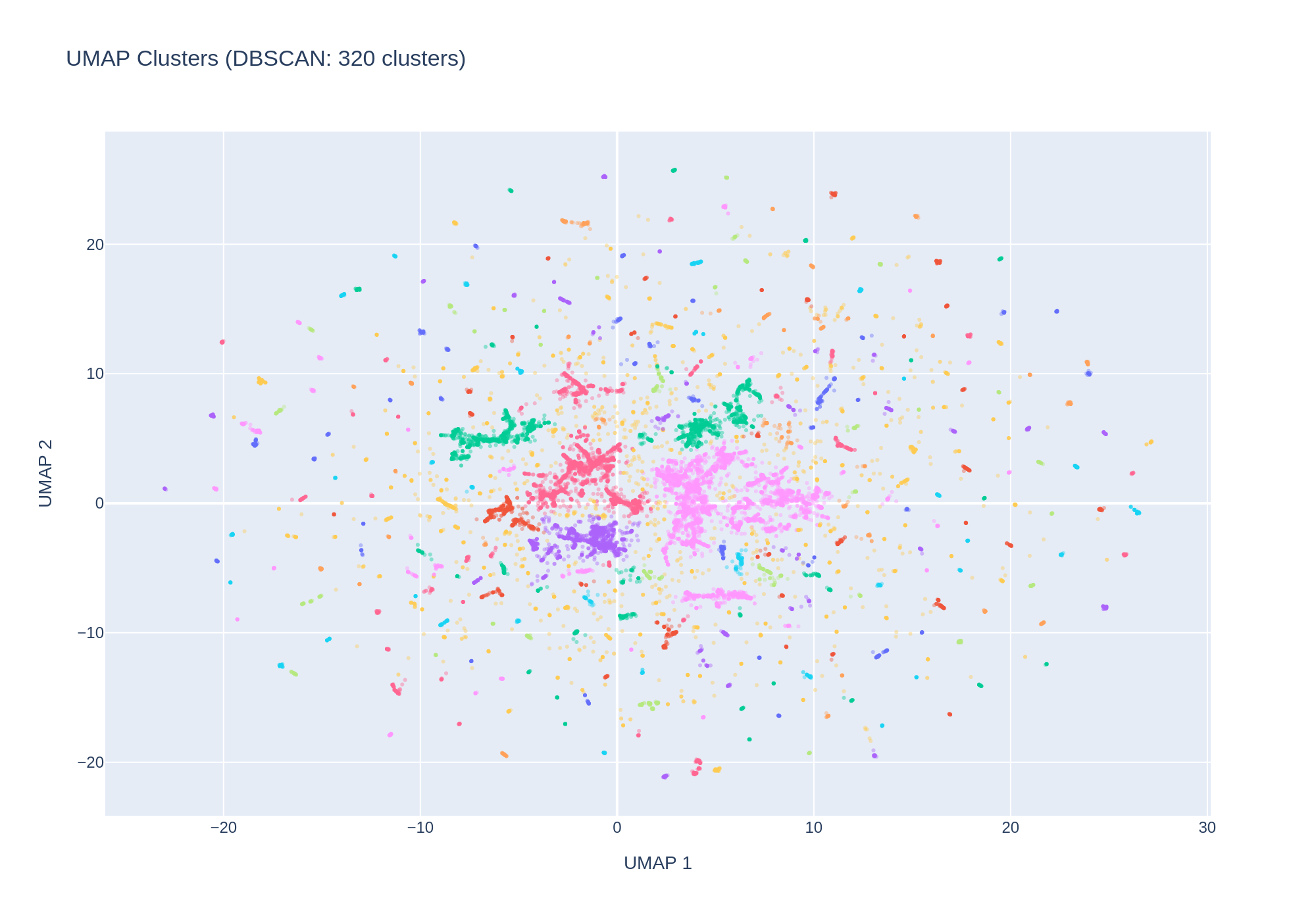
Umap Clusters
Notebooks
Data Files
| Filename | Size |
|---|---|
coverage_stats.csv |
0.2 KB |
isolation_source_raw_counts.csv |
174.7 KB |
ncbi_env_attribute_counts.csv |
6.7 KB |
umap_coords.csv |
3051.3 KB |