Web of Microbes Data Explorer
CompletedResearch Question
What does the kescience_webofmicrobes exometabolomics collection contain, which organisms overlap with the Fitness Browser, and how well do metabolite uptake/release profiles connect to pangenome-predicted metabolic capabilities?
Research Plan
Hypothesis
- H0: WoM metabolite profiles do not meaningfully connect to existing BERDL collections (low organism overlap, poor metabolite-to-pathway mapping)
- H1: WoM provides actionable cross-links — organisms overlap with the Fitness Browser, metabolites map to GapMind pathways and ModelSEED reactions, and exometabolomic profiles complement gene-level fitness data
Revision History
- v1 (2026-02-23): Initial plan — characterize WoM and assess cross-collection utility
Overview
Web of Microbes (WoM; Kosina et al. 2018, BMC Microbiology) is a curated exometabolomics database from the Northen lab at LBNL. It records which metabolites microorganisms consume (decrease) or produce (increase) when grown in defined environments. This project characterizes the BERDL-hosted copy (2018 archived snapshot): scale, organism coverage, metabolite composition, Fitness Browser overlap, and cross-collection linking potential to the pangenome and ModelSEED biochemistry.
Key Findings
1. WoM Action Encoding Uses Four Distinct Semantics, Not Three
The WoM database encodes metabolite observations with a 4-action system that differs between control and organism entries:
| Actor | Action | Meaning | Count |
|---|---|---|---|
| Control ("The Environment") | D | Detected in starting medium | 742 |
| Control ("The Environment") | N | Not detected | 1,023 |
| Organisms | I | Increased (present in medium, level went up) | 1,338 |
| Organisms | E | Emerged (NOT in medium, now detected = de novo production) | 1,155 |
| Organisms | N | No significant change | 7,509 |
'E' and 'I' are mutually exclusive (zero overlap across all 10,744 observations). 'E' encodes genuine de novo metabolite synthesis — the organism produced a metabolite that was absent from the starting medium. This distinction is biologically meaningful: 'I' represents amplification of existing metabolites, while 'E' represents novel biosynthetic output.
There is no "Decreased" (consumption) action for any organism in this 2018 snapshot. All 742 'D' observations belong exclusively to the control. The original Kosina et al. (2018) paper describes "decrease" as a valid action, so consumption data may exist in the newer GNPS2 version.
(Notebook: 01_database_overview.ipynb)
2. Two Direct Fitness Browser Strain Matches Plus Two Genus-Level Matches
| WoM Organism | FB Match | Match Type | FB Genes | FB Experiments |
|---|---|---|---|---|
| Pseudomonas sp. FW300-N2E3 | pseudo3_N2E3 |
Direct strain | 5,854 | 211 |
| Pseudomonas sp. GW456-L13 | pseudo13_GW456_L13 |
Direct strain | 5,243 | 106 |
| E. coli BW25113 | Keio |
Same strain | 4,610 | 168 |
| Synechococcus PCC7002 | SynE (PCC 7942) |
Genus only | 2,722 | 129 |
The two Pseudomonas direct matches are ENIGMA groundwater isolates with rich FB data. Keio (E. coli BW25113) is the same strain but WoM data is limited to 12 observations in ZMMG medium (sulfur metabolism focus only).
(Notebook: 01_database_overview.ipynb, 02_cross_collection_links.ipynb)
3. 19 WoM-Produced Metabolites Are Tested as FB Carbon/Nitrogen Sources
For pseudo3_N2E3, curated matching identified 19 metabolites that the organism produces (WoM) and that the Fitness Browser tests as carbon or nitrogen sources:
| WoM Metabolite | Action | FB Condition | FB Type |
|---|---|---|---|
| alanine | I | L-Alanine, D-Alanine | carbon/nitrogen |
| arginine | I | L-Arginine | nitrogen source |
| glycine | I | Glycine | nitrogen source |
| lactate | E | Sodium D-Lactate | carbon source |
| proline | I | L-Proline | carbon source |
| phenylalanine | I | L-Phenylalanine | carbon source |
| tryptophan | I | L-Tryptophan | nitrogen source |
| valine | E | L-Valine | carbon source |
| lysine | E | L-Lysine | nitrogen source |
| threonine | I | L-Threonine | nitrogen source |
| trehalose | I | D-Trehalose dihydrate | carbon source |
| adenine | I | Adenine hydrochloride | nitrogen source |
| adenosine | I | Adenosine | nitrogen source |
| inosine | I | Inosine | nitrogen source |
| thymine | E | Thymine | nitrogen source |
| malate | I | L-Malic acid | carbon source |
| nicotinamide | I | Nicotinamide | — |
| carnitine | E | Carnitine hydrochloride | — |
This enables the question: "Pseudomonas FW300-N2E3 produces lactate de novo (WoM action=E). Which genes are fitness-important for lactate utilization (FB)?" — directly connecting metabolite output to gene function. Of the 19 matches, 5 are de novo products (E) and 14 are amplified metabolites (I).
(Notebook: 02_cross_collection_links.ipynb)
4. 26.8% of WoM Metabolites Have Definitive ModelSEED Links (68.5% with Ambiguous Formula Matches)
| Match Type | WoM Compounds | Confidence | Percentage |
|---|---|---|---|
| Exact name match | 69 | High — 1:1 mapping | 26.8% |
| Formula-only match | 107 | Low — 1:8.4 avg expansion (107 WoM → 900 MS molecules) | 41.6% |
| Total with any link | 176 | Mixed | 68.5% |
| Unmatched | 81 | — | 31.5% |
Of 257 identified (non-unknown) WoM compounds, 69 (26.8%) have definitive ModelSEED links by exact name match. An additional 107 match by molecular formula alone, but formula matches are inherently ambiguous — each WoM formula maps to an average of 8.4 ModelSEED molecules (e.g., C5H11NO2 matches valine, norvaline, betaine, 5-aminopentanoate, and others). Formula matches provide candidate sets for manual curation, not definitive identifications.
(Notebook: 02_cross_collection_links.ipynb)
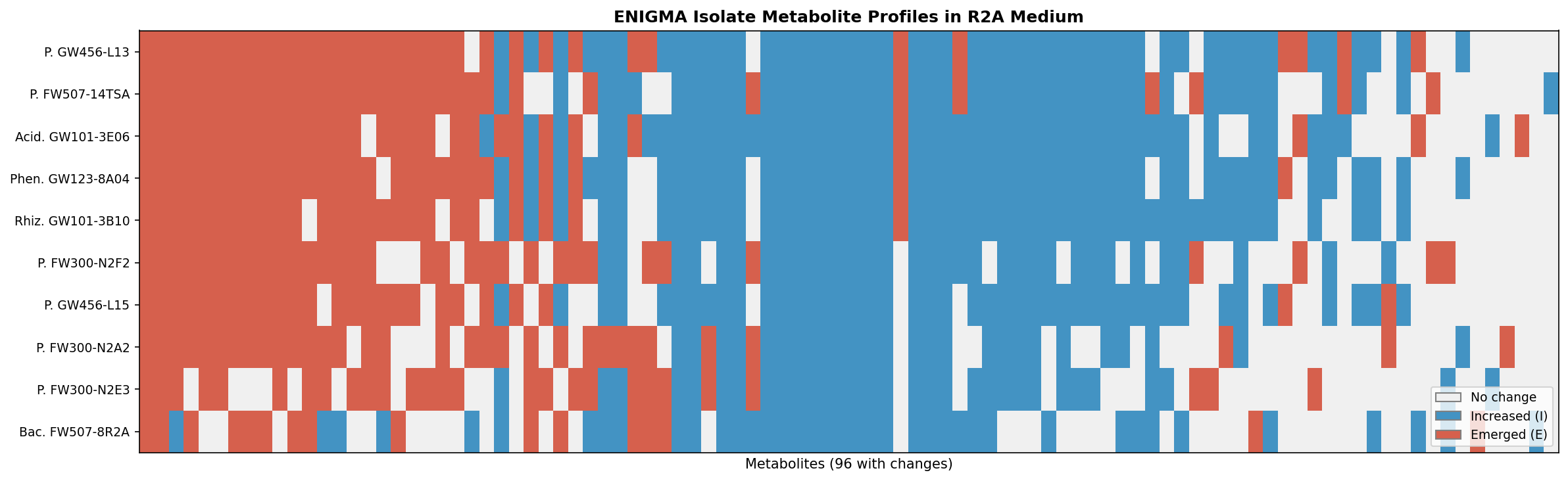
5. ENIGMA Isolates Show Distinct "Metabolic Novelty Rates"
The fraction of metabolite changes that represent de novo production ('E') versus amplification of existing metabolites ('I') varies 2-fold across ENIGMA isolates in R2A medium:
| Organism | Increased (I) | Emerged (E) | % Novel |
|---|---|---|---|
| Pseudomonas GW456-L13 | 49 | 34 | 32.4% |
| Pseudomonas FW507-14TSA | 44 | 33 | 31.4% |
| Pseudomonas FW300-N2A2 | 26 | 32 | 30.5% |
| Acidovorax GW101-3E06 | 48 | 30 | 28.6% |
| Bacillus FW507-8R2A | 39 | 16 | 15.2% |
GW456-L13 produces the most novel metabolites (32.4%), while FW507-8R2A produces the fewest (15.2%). This "metabolic novelty rate" is a potential phenotype for cross-referencing with pangenome gene content.
(Notebook: 01_database_overview.ipynb)
6. All WoM Genera Have Pangenome Species Clades
| Genus | Pangenome Species (top 5) | Total Genomes |
|---|---|---|
| Bacillus | 5 | 2,557 |
| Rhizobium | 5 | 449 |
| Pseudomonas fluorescens | 5 | 139 |
| Synechococcus | 5 | 88 |
| Phenylobacterium | 5 | 80 |
| Acidovorax | 5 | 79 |
| Zymomonas mobilis | 1 | 26 |
| Escherichia coli | 1 | 2 |
Pangenome context (gene clusters, conservation, functional annotations) is available for all WoM organism genera, enabling future genome-content ↔ metabolite-output analyses.
(Notebook: 02_cross_collection_links.ipynb)
Results
Database Scale
The 2018 WoM snapshot contains 37 organisms across 5 ENIGMA-funded projects:
- 6 biocrust isolates (5,604 observations in BG11 media) — the largest data block
- 10 ENIGMA groundwater isolates (1,050 observations in R2A medium)
- 4 other isolates (E. coli, Zymomonas, Synechococcus, Microcoleus)
- 10 triculture time-series entries and 2 native microbiome entries
- 4 theoretical auxotroph predictions and 1 control
589 metabolites are tracked, of which 332 (56.4%) are unidentified ("Unk_" prefix). Of the 257 identified metabolites, 408 (69.3% of all 589) show at least one change across the database. The most metabolically active compounds are amino acids (glutamine, proline, phenylalanine) and nucleotides (adenine, adenosine, guanine).
Cross-Collection Link Quality
WoM ↔ Fitness Browser: Strong for 3 organisms. The two Pseudomonas strains have >5,000 genes and >100 experiments each, with FB conditions that directly test metabolites WoM detects as produced. E. coli Keio is the same strain but WoM data is thin (12 observations, sulfur focus only).
WoM ↔ ModelSEED: Moderate. 26.8% of identified metabolites have definitive name-based links to ModelSEED molecules (high confidence). An additional 41.6% match by formula only (ambiguous, ~8:1 expansion ratio). Combined, 68.5% have at least one candidate link.
WoM ↔ GapMind: Blocked by naming convention mismatch. GapMind pathway names (internal IDs) don't contain simple metabolite names. A proper lookup table mapping GapMind pathways to substrate/product metabolites is needed.
WoM ↔ Pangenome: Available at genus level for all WoM organisms. Species-level matching requires strain-to-genome mapping (not attempted in this project).
Interpretation
Hypothesis Outcome
H1 is partially supported. Cross-collection links are real and actionable:
- 3 organisms have direct WoM ↔ FB connections with substantial fitness data
- 19 metabolites that N2E3 produces are tested as FB carbon/nitrogen sources
- 26.8% of identified metabolites have definitive ModelSEED links (68.5% with ambiguous formula matches)
- All genera have pangenome representation
However, the absence of consumption data is a fundamental limitation. The 2018 WoM snapshot records only what organisms produce, not what they consume. This prevents testing the most interesting question — whether consumed metabolites predict gene essentiality. The data is also small (37 organisms, 5 projects) and from a single laboratory.
Literature Context
- Kosina et al. (2018) introduced WoM with the same data we analyzed. Their paper describes "decrease" as a valid action, confirming that consumption data is part of the WoM schema but absent from this specific export.
- de Raad et al. (2022) developed the Northen Lab Defined Medium (NLDM) supporting growth of 108/110 phylogenetically diverse soil bacteria, with all metabolites trackable via LC-MS/MS. This represents a major expansion of the exometabolomics approach and likely contains consumption data. The NLDM dataset may be available in the current GNPS2 version of WoM.
- Jacoby & Kopriva (2019) reviewed exometabolomics as a tool for understanding microbial cross-feeding and resource competition, noting its complementarity with genomics data — precisely the integration this project assessed.
- Douglas (2020) framed the microbial exometabolome as an "ecological resource and architect of microbial communities," highlighting the need for databases linking exometabolomics to genomics.
Novel Contribution
This project provides the first characterization of WoM data within a multi-collection lakehouse context. Key novel findings:
- Action encoding clarification: The E/I distinction (emerged vs increased) was not prominently documented and required data exploration to decode. This is critical for correct biological interpretation.
- Cross-collection link assessment: Quantified that 26.8% of WoM metabolites have definitive ModelSEED links, 19 metabolites bridge directly to FB carbon/nitrogen source experiments, and 3 organisms span both collections — demonstrating that metabolite-to-gene integration is technically feasible.
- Metabolic novelty rate: The 15–32% range of de novo production across ENIGMA isolates is a new phenotypic measure enabled by the E/I distinction.
Limitations
- No consumption data: The most scientifically valuable WoM data (what organisms consume) is absent from this snapshot.
- Small organism set: 37 organisms (20 experimental) is too few for statistical analyses across organism groups.
- 2018 frozen snapshot: The Wayback Machine archive may not reflect the current state of WoM. The GNPS2-hosted version or the Northen lab's internal datasets (including the NLDM 110-organism panel from de Raad et al. 2022) would be substantially richer.
- GapMind pathway matching requires a lookup table: Simple name matching failed. A dedicated pathway-to-metabolite mapping table would enable this link.
- WoM E. coli data is minimal: Despite Keio being one of the richest FB organisms, WoM has only 12 observations for E. coli BW25113 (sulfur/cysteine focus).
Future Directions
- Obtain the current WoM dataset from GNPS2 or the Northen lab: The de Raad et al. (2022) NLDM study tested 110 soil bacteria with full consumption and production tracking. This dataset, if ingested, would resolve the no-consumption limitation and increase the organism count 3–5×.
- Build a GapMind pathway ↔ metabolite lookup table: Map each GapMind pathway to its substrate and product metabolites using the GapMind documentation, enabling the WoM ↔ GapMind link that failed in NB02.
- Gene fitness ↔ metabolite production analysis: For pseudo3_N2E3 and pseudo13_GW456_L13, identify which genes are fitness-important on carbon sources that the same organism produces. This tests whether metabolic output predicts gene essentiality.
- Metabolic novelty rate ↔ pangenome openness: Test whether organisms with higher E/(E+I) ratios (more de novo production) have more open pangenomes or more accessory genes.
- Re-ingest with consumption data: When a newer WoM dataset is available, re-run the ingestion pipeline (
data/wom_ingest/) to updatekescience_webofmicrobesand enable the consumption-based analyses that are currently blocked.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kescience_webofmicrobes |
compound, environment, organism, project, observation |
Exometabolomics data (metabolite production/excretion profiles) |
kescience_fitnessbrowser |
organism, gene, experiment |
Fitness data for organism overlap and condition matching |
kbase_msd_biochemistry |
molecule |
Compound name/formula matching for reaction-level annotation |
kbase_ke_pangenome |
pangenome, gapmind_pathways |
Species clade counts and pathway predictions |
Generated Data
| File | Rows | Description |
|---|---|---|
data/wom_organisms.csv |
37 | WoM organisms with observation counts and category labels |
data/wom_compounds.csv |
589 | WoM metabolites with action counts and chemical class |
data/wom_environments.csv |
10 | Growth media with organism/observation counts |
data/wom_projects.csv |
5 | ENIGMA projects with DOIs |
data/fb_overlap.csv |
2 | Direct WoM ↔ FB strain matches |
data/modelseed_name_matches.csv |
75 | WoM ↔ ModelSEED exact name matches |
data/modelseed_formula_matches.csv |
900 | WoM ↔ ModelSEED formula matches |
References
- Kosina SM, Greiner AM, Lau RK, Jenkins S, Baran R, Bowen BP, Northen TR. (2018). "Web of microbes (WoM): a curated microbial exometabolomics database for linking chemistry and microbes." BMC Microbiology, 18:139. PMID: 30208844
- de Raad M, Li YV, Kuehl JV, Andeer PF, Kosina SM, Hendrickson A, Saichek NR, Golini AN, Han Z, Wang Y, Bowen BP, Deutschbauer AM, Arkin AP, Chakraborty R, Northen TR. (2022). "A Defined Medium for Cultivation and Exometabolite Profiling of Soil Bacteria." Front Microbiol, 13:855331. PMID: 35694313
- Douglas AE. (2020). "The microbial exometabolome: ecological resource and architect of microbial communities." Phil Trans R Soc B, 375:20190250.
- Jacoby RP, Kopriva S. (2019). "Metabolic niches in the rhizosphere microbiome: new tools and approaches to analyse metabolic mechanisms of plant-microbe nutrient exchange." J Exp Bot, 70:697-713. PMID: 30576534
- Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, Kuehl JV, Melnyk RA, Lamson JS, Cai Y, Carlson HK, Bristow J, Arkin AP, Deutschbauer AM. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature, 557:503-509.
Data Collections
Review
Summary
This is a well-executed exploratory data characterization project that inventories the kescience_webofmicrobes exometabolomics collection and systematically assesses its cross-collection linking potential to the Fitness Browser, ModelSEED biochemistry, GapMind pathways, and the pangenome. The project excels in documentation quality: the three-file structure (README, RESEARCH_PLAN, REPORT) is thorough and internally consistent, with clear hypotheses, honest limitation reporting, and actionable future directions. Both notebooks are executed with saved outputs, produce reproducible intermediate data, and follow good Spark-first data access patterns. The six key findings — particularly the E/I action encoding clarification, the identification of 19 curated metabolite-condition overlaps for pseudo3_N2E3, and the 26.8% definitive ModelSEED coverage — are well-supported by the data shown. The main areas for improvement are the single-figure output (more visualizations would strengthen the narrative), the completely failed GapMind pathway mapping (0/20 amino acids matched), and the absence of the planned third notebook, though this deferral is well-documented and justified.
Methodology
Research question: Clearly stated and testable. The three-part question (what does WoM contain, which organisms overlap with FB, how do metabolite profiles connect to pangenome capabilities?) is well-scoped and decomposed into two focused notebooks.
Hypothesis: Well-formulated with explicit H0/H1 and measurable criteria. The REPORT honestly concludes "H1 is partially supported" — a nuanced outcome that acknowledges both the real cross-links discovered and the fundamental limitation of absent consumption data.
Data sources: All four BERDL collections (kescience_webofmicrobes, kescience_fitnessbrowser, kbase_msd_biochemistry, kbase_ke_pangenome) are clearly identified in the README, RESEARCH_PLAN, and REPORT, with table names, estimated row counts, and filter strategies documented upfront. The query strategy table in RESEARCH_PLAN is a valuable practice.
Approach soundness: The overall approach is appropriate — inventory first (NB01), then cross-collection linking (NB02). The Fitness Browser organism matching uses a reasonable two-stage strategy (exact strain ID via regex extraction and normalization, then genus-level fallback for non-ENIGMA organisms). The ModelSEED matching (exact name first, formula for remainder) is methodical and results are clearly separated by confidence level.
Reproducibility: Someone with BERDL JupyterHub access could reproduce this analysis. The README includes a reproduction section with a clear dependency diagram (NB01 → data/*.csv → NB02 → data/modelseed_*.csv), notes that both notebooks require active Spark sessions, and explains why NB03 was deferred. All intermediate data files are saved as CSVs. A requirements.txt with version constraints (pandas>=1.5,<3.0, pyspark>=3.4,<4.0, matplotlib>=3.5,<4.0) is provided. Estimated runtimes are not documented but would be short given the small WoM table sizes.
Code Quality
SQL queries: Correct and well-structured. NB01's organism-observation join uses appropriate LEFT JOIN with aggregation and action-count pivoting. NB02's ModelSEED matching is methodical. The pangenome genus search in NB02 uses LIKE patterns (e.g., LIKE '%Pseudomonas%fluorescens%'), which docs/pitfalls.md advises against for performance — but this is acceptable for these small exploratory queries against the pangenome summary table.
Metabolite-condition matching (NB02, cell fb-wom-metabolite-conditions): Uses a well-curated dictionary mapping (curated_map) that maps WoM metabolite names to their specific FB condition variants (e.g., "alanine" → ["L-Alanine", "D-Alanine"]). This produces 19 high-confidence matches with clear biological relevance. This is a strength — the curated approach avoids the false-positive inflation that naive substring matching would produce.
Notebook organization: Both notebooks follow a clean setup → query → analysis → save structure with numbered markdown section headers. NB01 includes a summary cell at the end recapping key counts. Code is readable with inline comments explaining non-obvious logic (e.g., the categorize_organism function, the classify_metabolite function). One minor issue: NB01 section numbering jumps from "## 7" to "## 9. Save Outputs," with the heatmap section labeled "## 8" appearing after section 9 in the cell order — apparently the heatmap cell was inserted after the save cell.
Pitfall awareness: The RESEARCH_PLAN explicitly notes "FB columns are all strings (CAST needed)" and "GapMind requires MAX aggregation per genome-pathway pair" from docs/pitfalls.md. The first is not exercised in the analysis (no numeric comparisons on FB columns), and the second becomes moot because GapMind matching failed at the name-lookup stage before aggregation was needed. The CAST(no_genomes AS INT) in NB02's pangenome query correctly handles the string-typed numeric columns. No .toPandas() calls are made on large tables — all heavy filtering happens in Spark SQL first, consistent with pitfall guidance about unnecessary .toPandas() calls.
GapMind matching failure (NB02, cells gapmind-pathways and gapmind-carbon): The approach of searching GapMind pathway names for substring matches with amino acid names (e.g., looking for "L-arginine" in pathway names) yields 0/20 matches. This is because GapMind pathway IDs in the kbase_ke_pangenome.gapmind_pathways table use internal identifiers that don't contain simple metabolite substrings. The notebook correctly identifies this as a blocking issue ("Needs a pathway-to-metabolite lookup table"), but the approach could have been validated earlier by inspecting sample pathway names before writing the full matching loop. The cell output clearly shows the failure, which is honest reporting.
Minor code issues:
- NB01 cell organisms-detail: The display() call on a string (from .to_string()) wraps the output in quotes, producing repr-style output. Using print() would be cleaner.
- NB02 cell integration-test: This cell describes what could be done with the cross-collection links but doesn't execute any actual integration query. It serves as a narrative summary, which is useful documentation, but the section title "Integration test" implies computation that doesn't occur.
Findings Assessment
Finding 1 (Action encoding): Well-supported. The observation data clearly shows 0 'D' actions for any organism (all 742 'D' belong to the control), and mutual exclusivity of 'E' and 'I'. The biological distinction between "emerged" (de novo production) and "increased" (amplification of existing) is genuinely useful and not prominently documented in the original Kosina et al. (2018) paper. This is the project's most novel contribution.
Finding 2 (FB matches): Accurate. Two direct strain matches (pseudo3_N2E3, pseudo13_GW456_L13) confirmed by strain ID normalization, plus E. coli BW25113/Keio (same strain, thin WoM data) and Synechococcus PCC7002/SynE (genus-level only, different strain). The match table in the REPORT correctly distinguishes "Direct strain" from "Same strain" and "Genus only" match types.
Finding 3 (19 metabolite-condition overlaps): The curated table of 19 matches is biologically meaningful and well-presented. The mix of de novo products (E: lactate, valine, lysine, thymine, carnitine) and amplified metabolites (I: amino acids, nucleotides) is clearly shown. The connection to specific FB experimental conditions (carbon source vs nitrogen source) is actionable for future work.
Finding 4 (ModelSEED matching — 26.8% name, 68.5% total): Solid and clearly presented. The REPORT appropriately distinguishes high-confidence name matches (69 compounds, 1:1 mapping) from ambiguous formula-only matches (107 compounds → 900 ModelSEED molecules, ~8.4:1 expansion). The 31.5% unmatched rate is honestly reported. The REPORT could benefit from explicitly noting that formula matches are candidate sets rather than definitive identifications — this nuance is present but could be more prominent.
Finding 5 (Metabolic novelty rates): An interesting and creative metric. The E/(E+I) ratio varying from 15.2% (Bacillus FW507-8R2A) to 32.4% (Pseudomonas GW456-L13) across ENIGMA isolates in R2A medium is a novel phenotypic characterization. No statistical tests are applied (appropriate given n=10 organisms and single-environment data), and no overinterpretation is attempted.
Finding 6 (Pangenome coverage): Confirmed at genus level for all WoM organisms. The caveat that species-level matching requires strain-to-genome mapping (not attempted) is appropriate.
Limitations: Thoroughly and honestly documented. The five limitations in the REPORT are specific and actionable: (1) no consumption data, (2) small organism set, (3) 2018 frozen snapshot, (4) GapMind naming mismatch, (5) minimal E. coli WoM data. The pointer to de Raad et al. (2022) NLDM dataset for newer data is valuable context.
NB03 deferral: The RESEARCH_PLAN described 3 notebooks, with NB03 marked "if warranted." The README explicitly documents the deferral with a clear justification: "The absence of consumption data in this 2018 WoM snapshot made organism-comparison heatmaps less informative than planned." This is honest and well-handled.
Suggestions
-
[Moderate] Add more visualizations: The project has a single figure (the ENIGMA metabolite heatmap, which is effective). Additional figures would strengthen the narrative: (a) a bar chart of metabolite categories (amino acids, nucleotides, etc.) showing the category breakdown from NB01; (b) a Venn or UpSet diagram showing cross-collection overlap (how many organisms/metabolites are in WoM only, WoM+FB, WoM+ModelSEED, etc.); (c) a scatter plot of emerged-vs-increased counts per organism showing the metabolic novelty rate variation. The REPORT references these findings textually but visualizations would make them more immediately accessible.
-
[Moderate] Investigate GapMind pathway naming before writing full matching loop: The 0/20 failure in NB02 could have been caught earlier by inspecting a few sample pathway names (e.g.,
SELECT DISTINCT pathway FROM kbase_ke_pangenome.gapmind_pathways LIMIT 10). The notebook does show this query but the matching code was written before understanding the naming convention. A brief note in the notebook explaining what the pathway names look like and why simple substring matching fails (they use internal pathway IDs like compound utilization pathway names, not "L-arginine biosynthesis") would help future researchers avoid the same dead end. -
[Moderate] Add estimated runtimes to reproduction section: Both notebooks appear to run quickly (WoM tables are tiny, and the cross-collection queries are well-filtered), but documenting approximate runtimes (e.g., "NB01: ~2 minutes, NB02: ~3 minutes") would help prospective reproducers plan their sessions.
-
[Minor] Fix NB01 section numbering: The heatmap section (labeled "## 8") appears after the save section (labeled "## 9. Save Outputs") in the cell order. Either reorder the cells or renumber the sections for sequential flow.
-
[Minor] Consider adding seaborn or scipy to requirements.txt: If additional visualizations are added per suggestion 1,
seabornand/orscipywould likely be needed and should be pinned inrequirements.txt. Currently onlypandas,pyspark, andmatplotlibare listed, which is correct for the current code. -
[Minor] Expand the E. coli / Keio analysis: The REPORT notes that E. coli BW25113 has only 12 WoM observations (sulfur/cysteine focus), making it a weak cross-link. NB02 could briefly quantify this — how many E. coli metabolites overlap with Keio FB conditions? Even a negative result ("only 1-2 overlaps due to sulfur-only focus") would strengthen the claim that the Pseudomonas matches are the primary integration opportunity.
-
[Nice-to-have] Explore GapMind pathway names more deeply: The
gapmind_pathways.pathwaycolumn contains 80 distinct pathway names. Printing all 80 in the notebook (rather than just the LIMIT 200 query) and categorizing them by type (amino acid utilization, carbon source utilization, etc.) could reveal which pathways are matchable to WoM metabolites, even if the simple substring approach failed. Some GapMind pathways may use metabolite names in non-obvious formats (e.g., "D-glucose utilization" rather than just "glucose").
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Enigma Metabolite Heatmap