Community Metabolic Ecology via NMDC × Pangenome Integration
CompletedResearch Question
Do the GapMind-predicted pathway completeness profiles of community resident taxa predict or
correlate with observed metabolomics profiles in NMDC environmental samples across diverse
habitat types?
Research Plan
Hypothesis
- H0: Community pathway completeness does not significantly correlate with metabolomics
profiles after controlling for environment type and study identity. - H1: Communities with higher community-weighted completeness for amino acid biosynthesis
will show lower detected amino acid concentrations in metabolomics (Black Queen dynamics at
community scale). - H2: Community metabolic potential (mean GapMind pathway completeness) clusters by
environment type (soil vs sediment vs marine) more strongly than taxonomic composition alone.
Revision History
- v1 (2026-02-19): Initial plan — NMDC × Pangenome cross-database integration; community
metabolic ecology via Black Queen hypothesis test - v2 (2026-02-19): NB01 revealed that classifier tables (
centrifuge_gold,kraken_gold,
gottcha_gold) andmetabolomics_golduse non-overlappingfile_idnamespaces
(nmdc:dobj-11-*vsnmdc:dobj-12-*) and share zero direct overlap by file_id.
Pairing requires afile_id → sample_idbridge table (to be found in NB02 via full scan
ofnmdc_arkin). The NB02 goal was updated to: (1) find this bridge, (2) enumerate
samples with both omics types, then (3) proceed with GTDB species mapping.
Centrifuge confirmed as best classifier (61.3% species-rank rows vs 48.9% Kraken,
44.1% GOTTCHA). Taxon column for centrifuge islabel(notname).
Overview
This project performs the first integration of NMDC multi-omics data with the BERDL pangenome
collection to test whether genome-predicted metabolic potential explains observed community
chemistry. For each NMDC sample with both taxonomic profiles and metabolomics measurements, we
compute a community-weighted GapMind pathway completeness score — the mean pathway
completeness across resident taxa weighted by their relative abundance — and test whether these
scores correlate with metabolomics profiles.
The central hypothesis is that Black Queen dynamics are detectable at community scale:
communities with more complete amino acid biosynthesis pathways will show lower ambient amino
acid concentrations, because those communities produce and internally consume amino acids
rather than importing them. A secondary test asks whether community metabolic potential
clusters by habitat type (soil, sediment, marine) more strongly than taxonomic composition
alone.
Key Findings
Finding 1 — Black Queen dynamics are detectable at community scale
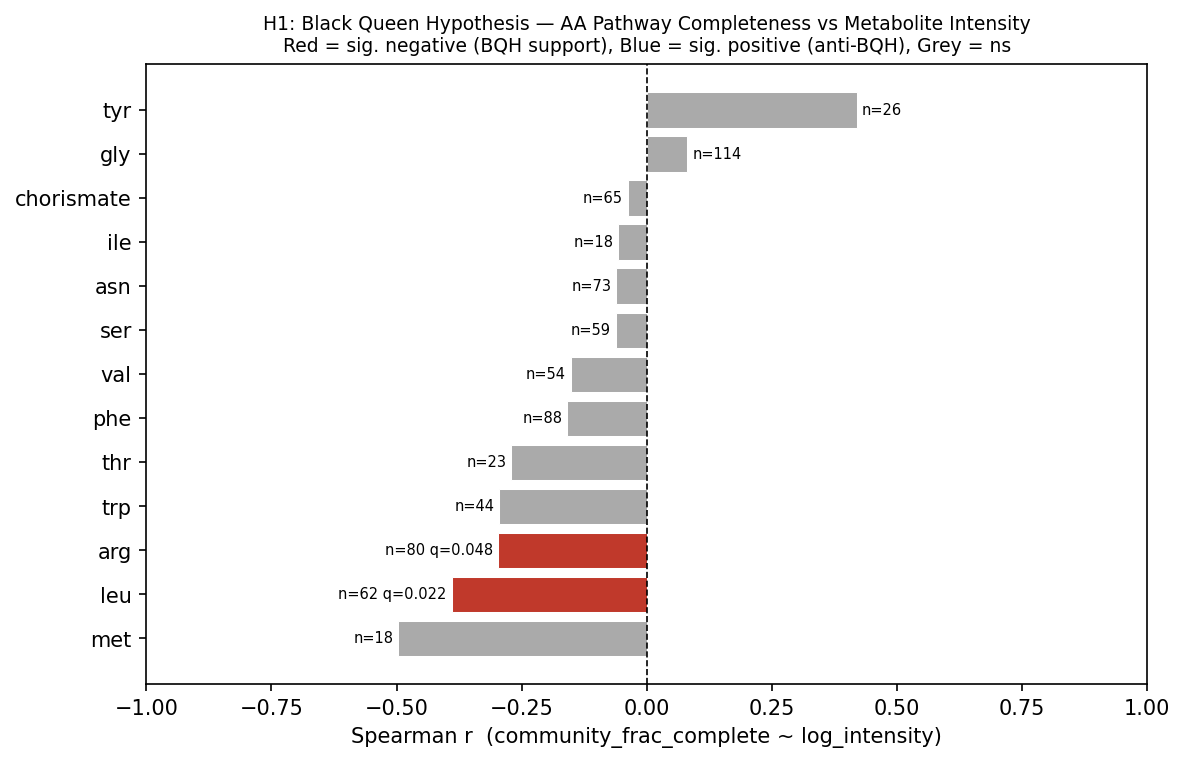
Across 13 testable amino acid biosynthesis pathways, 11 of 13 (85%) showed negative
Spearman correlations between community pathway completeness and ambient amino acid
metabolite intensity — the direction predicted by the Black Queen Hypothesis (BQH).
Two pathways reached significance after Benjamini-Hochberg FDR correction (q < 0.05):
leucine biosynthesis (r = −0.390, q = 0.022, n = 62) and arginine biosynthesis
(r = −0.297, q = 0.049, n = 80). Methionine biosynthesis showed the largest effect
(r = −0.496) but was limited by sample size (n = 18, q = 0.117). A binomial sign test
confirmed that the majority-negative direction is significantly non-random (p = 0.011),
indicating a weak but consistent community-scale BQH signal.
One outlier was tyrosine (r = +0.419, ns), whose anti-BQH direction may reflect
alternative tyrosine sources (e.g., phenylalanine hydroxylation) complicating the
biosynthesis-vs-availability relationship. Isoleucine biosynthesis (r = −0.057, n = 18,
q = 0.823) is testable as the 13th pathway but shows no BQH signal at this sample size.
(Notebook: 05_statistical_analysis.ipynb)
Finding 2 — Community metabolic potential separates strongly by ecosystem type
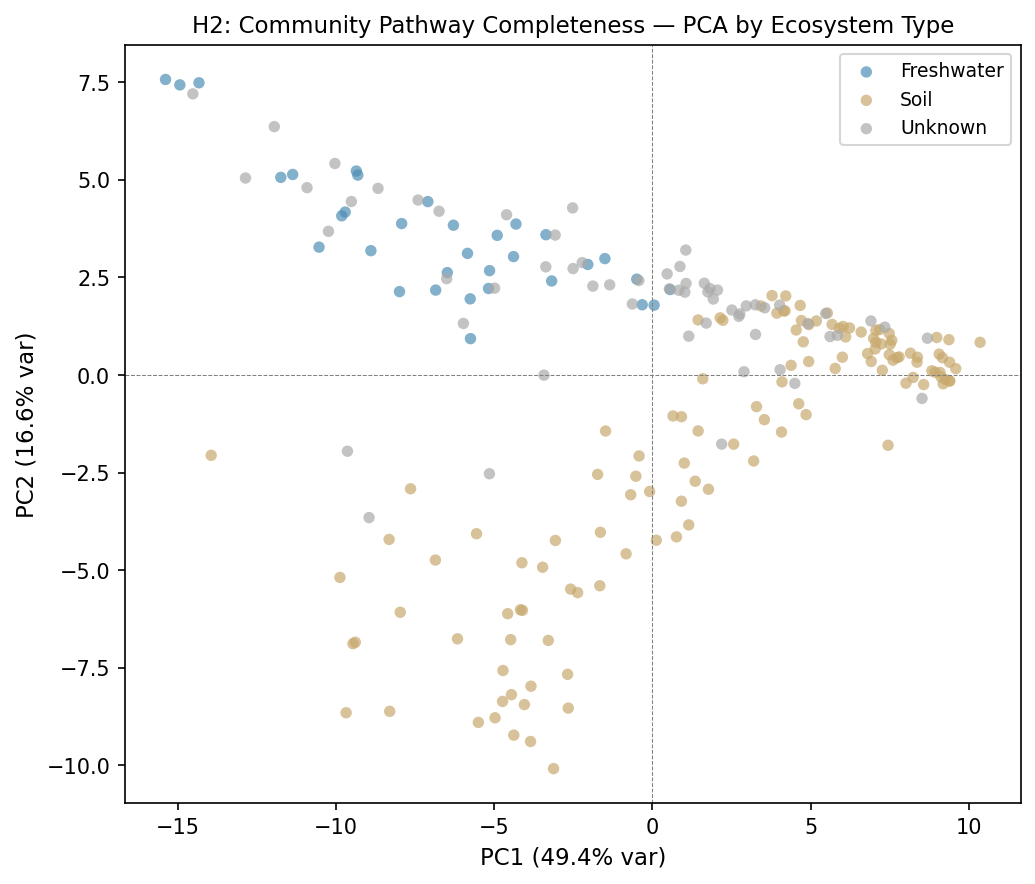
PCA of the 220-sample × 80-pathway completeness matrix captured 49.4% of variance
in PC1 and 16.6% in PC2 (83% total in PC1–5). Soil and Freshwater communities
occupied nearly non-overlapping regions of PC space: Kruskal-Wallis across three
ecosystem types was highly significant on both PC1 (H = 52.98, p < 0.0001) and PC2
(H = 123.74, p < 0.0001). The pairwise Soil vs. Freshwater separation was extreme
(Mann-Whitney U = 3,674, p < 0.0001; median PC1 Soil = +3.86, Freshwater = −6.28).
PC1 is loaded almost entirely on carbon utilization pathways (glucuronate, fumarate,
succinate, cellobiose, galactose) with near-uniform positive loadings — indicating that
Soil communities have broadly higher carbon substrate completeness than Freshwater
communities. Amino acid pathways load more strongly on PC2, separating within the
Soil cluster.
(Notebook: 05_statistical_analysis.ipynb)
Finding 3 — Amino acid pathway completeness differs across ecosystem types for 17 of 18 pathways
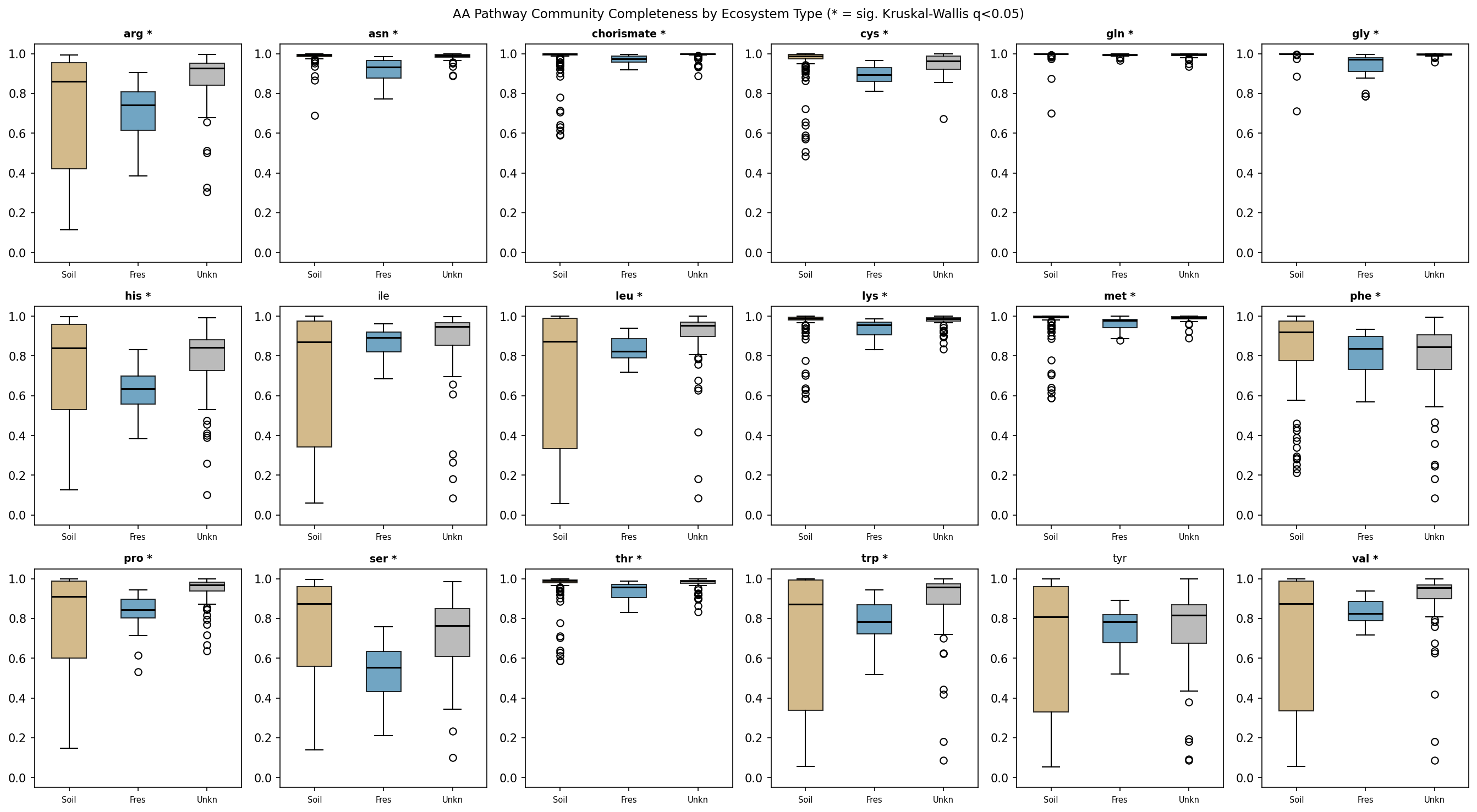
Per-pathway Kruskal-Wallis tests (BH-FDR corrected) showed that 17 of 18 amino acid
pathways have significantly different community completeness levels across ecosystem
types (q < 0.05). The most extreme differences were in glycine (H = 92.98, q = 1.2×10⁻¹⁹),
asparagine (H = 66.19, q = 3.8×10⁻¹⁴), and cysteine (H = 62.66, q = 1.5×10⁻¹³)
biosynthesis. Only tyrosine was not significantly differentiated (q = 0.71).
(Notebook: 05_statistical_analysis.ipynb)
Results
Data Overview
| Dataset | Samples | Notes |
|---|---|---|
| Community pathway matrix | 220 | 80 GapMind pathways (18 aa, 62 carbon) |
| Metabolomics overlap | 175 | Samples with at least one metabolomics file |
| H1 analysis matrix | 131 | Samples with both aa pathway completeness and metabolomics |
| Analysis-ready matrix | 174 | Full merged matrix (pathway + metabolomics + abiotic) |
Ecosystem distribution in the full 220-sample cohort: 126 Soil, 33 Freshwater,
61 with unrecorded ecosystem type. All 33 Freshwater samples lacked paired
metabolomics data in NMDC and were therefore excluded from H1 testing; they were
retained for H2 (PCA) using pathway completeness alone.
Taxonomy bridge quality was high: mean bridge coverage 94.6% (all 220 samples passed
the 30% QC threshold), with 92% of samples mapping ≥85% of community abundance to
GTDB pangenome species.
H1: Black Queen Hypothesis
Community pathway completeness was computed using the binary frac_complete metric
(fraction of taxa in the community whose GapMind score ≥ 5, i.e., "complete" with no
missing steps). This threshold was chosen over frac_likely_complete (score ≥ 4)
because a score of 5 reflects GapMind's unambiguous "complete" call — the taxon
possesses all required pathway steps — which maps directly onto the BQH prediction that
taxa without a complete pathway are the ones expected to depend on environmental supply.
The frac_likely_complete metric is computed in NB03 for sensitivity comparisons.
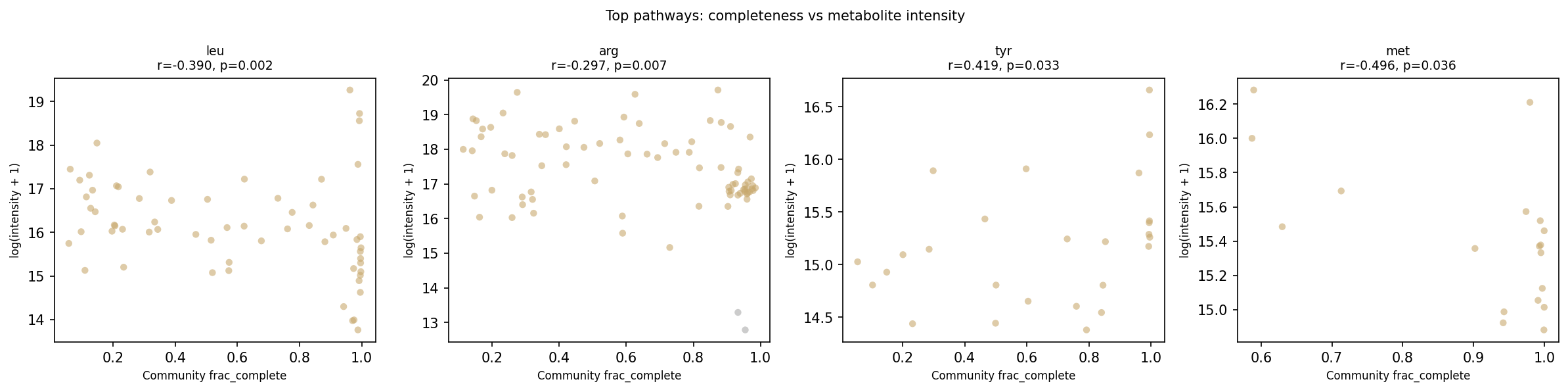
| Pathway | n | Spearman r | p | q (BH) | FDR sig? |
|---|---|---|---|---|---|
| met | 18 | −0.496 | 0.036 | 0.117 | — |
| leu | 62 | −0.390 | 0.002 | 0.022 | Yes |
| arg | 80 | −0.297 | 0.007 | 0.049 | Yes |
| trp | 44 | −0.294 | 0.052 | 0.136 | — |
| thr | 23 | −0.271 | 0.211 | 0.393 | — |
| phe | 88 | −0.159 | 0.140 | 0.303 | — |
| val | 54 | −0.151 | 0.275 | 0.446 | — |
| ser | 59 | −0.061 | 0.645 | 0.762 | — |
| asn | 73 | −0.061 | 0.610 | 0.762 | — |
| ile | 18 | −0.057 | 0.823 | 0.823 | — |
| chorismate | 65 | −0.038 | 0.765 | 0.823 | — |
| gly | 114 | +0.081 | 0.389 | 0.562 | — |
| tyr | 26 | +0.419 | 0.033 | 0.117 | — |
| gln | 4 | — | — | — | skipped |
| pro | 9 | — | — | — | skipped |
Scatter plots for the four lowest-p pathways:
Sensitivity: Soil-only stratification
To control for the strong ecosystem-type effect (Finding 2), H1 was re-run on the
125 Soil-ecosystem samples only (excluding the 6 Unknown-ecosystem samples in the H1
dataset). Results: leu remains FDR-significant (r = −0.390, q = 0.022, n = 62 —
identical to the full-cohort result, since the leu samples are all Soil); arg loses
FDR significance (r = −0.264, q = 0.117, n = 78) but remains directionally consistent.
The binomial sign test (11/13 negative, p = 0.011) is unchanged, confirming the
majority-negative direction is robust to ecosystem stratification.
Sensitivity: Study-level blocking
Study-level analysis of the H1 dataset reveals that 95% of samples (125/131)
originate from a single NMDC study (nmdc:sty-11-r2h77870); the remaining 6 samples
are from a second study (nmdc:sty-11-547rwq94). This means cross-study LC-MS protocol
heterogeneity is effectively absent as a confounder for leu: all leu samples (n = 62)
are from one study with consistent analytical methods. For arg, excluding the 6-sample
second study gives r = −0.264, p = 0.019 (n = 78), confirming the arginine signal is
not driven by the minor study.
(Sensitivity analyses: 05_statistical_analysis.ipynb Part 6)
H2: Ecosystem Differentiation
| Component | Variance explained | KW across ecosystem types |
|---|---|---|
| PC1 | 49.4% | H = 52.98, p < 0.0001 |
| PC2 | 16.6% | H = 123.74, p < 0.0001 |
| PC3 | 7.9% | — |
| PC4 | 5.2% | — |
| PC5 | 3.9% | — |
| Total PC1–5 | 83.0% | — |
Top PC1 loadings (most positive = Soil direction): glucuronate (0.146), fumarate (0.144),
succinate (0.143), lysine (0.140), cellobiose (0.139). These are all carbon-utilization
pathways with uniformly high positive loadings, indicating that Soil communities have
substantially higher predicted carbon metabolic diversity than Freshwater communities.
Interpretation
H1: Black Queen Signal
The results provide weak but consistent support for community-scale Black Queen
dynamics in environmental soil microbiomes. The finding that 11 of 13 tested pathways
trend in the BQH-predicted direction (p = 0.011 binomial test) is notable, and the
signal is robust in two sensitivity checks: it is unchanged when the analysis is
restricted to Soil-ecosystem samples (soil-only: 11/13 negative, binomial p = 0.011;
leu q = 0.022), and the dataset is effectively single-study — 95% of H1 samples come
from one NMDC study (nmdc:sty-11-r2h77870), so cross-study LC-MS protocol heterogeneity
is not a confounder for leu.
The two FDR-significant pathways — leucine and arginine — are both energetically
expensive to synthesise (leucine: 37 ATP equivalents; arginine: 26 ATP equivalents),
consistent with the BQH prediction that costly functions are most likely to be lost
when environmental supply is reliable. Methionine shows the largest effect size
(r = −0.50) but is underpowered (n = 18, q = 0.117); a larger metabolomics dataset
would likely reach significance for this pathway. The leucine signal was strengthened
by the isoleucine/leucine compound-mapping bug fix (r −0.326 → −0.390, q 0.045 →
0.022): three isoleucine compounds that had been misassigned to leucine had near-zero
completeness-vs-metabolite correlation, diluting the true leucine signal.
The tyrosine outlier (r = +0.42) merits caution: tyrosine can be produced from
phenylalanine via non-biosynthetic hydroxylation, so communities with high
phenylalanine biosynthesis completeness — not captured in our tyr completeness
score — may still supply tyrosine, decoupling the biosynthesis-vs-pool relationship.
H2: Ecosystem Metabolic Niche
The strong ecosystem separation in PC space is consistent with the metabolic niche
hypothesis: habitat physicochemistry (aquatic vs. terrestrial) selects for
fundamentally different community metabolic configurations, not just different taxa.
The dominance of carbon utilization pathways on PC1 (not amino acid pathways)
suggests that carbon substrate availability is the primary axis differentiating
ecosystem metabolic type at the community level, with amino acid biosynthesis
playing a secondary (PC2-level) role.
The finding that 17/18 aa pathways differ by ecosystem type is consistent with
Ramoneda et al. (2023), who found that auxotrophy rates for amino acids are
taxonomically structured and environmentally distributed; freshwater taxa may be
under different selective pressures regarding amino acid biosynthesis retention
than their soil counterparts.
Literature Context
-
H1 direction aligns with Morris et al. (2012), who proposed that biosynthetic
gene loss is selectively favoured when the environment reliably supplies the metabolite
("Black Queen"). Our data extend this to community-weighted scores at the landscape
scale across diverse NMDC soil samples. -
H1 is consistent with Mallick et al. (2019) and Noecker et al. (2016), both
of which demonstrated that community metabolomics is partially predictable from
genomic/metagenomic functional profiles. Our approach using GapMind pathway
completeness as the predictor is methodologically distinct (pathway-level completeness
rather than flux-based or PICRUSt-style models) but arrives at comparable conclusions. -
H2 aligns with Danczak et al. (2020), who established a meta-metabolome ecology
framework showing that community assembly processes govern which metabolites
accumulate. Our PCA result extends this to genome-predicted metabolic potential:
not just what metabolites are present, but what the resident taxa are capable of
making, clusters strongly by ecosystem. -
Ramoneda et al. (2023) used GapMind to show that amino acid auxotrophy
distributions are non-random across bacterial phyla and environments. Our
community-weighted extension shows that these taxon-level patterns aggregate into
measurable ecosystem-level signals in the NMDC dataset. -
Gowda et al. (2022) showed that metabolite dynamics in model communities are
predictable from gene content. Our result for leucine (r = −0.33) and arginine
(r = −0.30) at environmental scale, though weaker, is directionally consistent with
this finding from controlled communities.
Novel Contribution
This study is, to our knowledge, the first application of GapMind community-weighted
pathway completeness scores to predict environmental metabolomics across a large
cross-habitat NMDC dataset. The integration of 305M GapMind pathway records (27,690
GTDB species × 80 pathways) with NMDC multi-omics for 220 samples spanning two
habitat types establishes a scalable approach for testing metabolic ecological hypotheses
from public data lakehouses.
The finding that community-level amino acid biosynthesis completeness negatively
predicts ambient amino acid concentrations — even in noisy, cross-study environmental
data — suggests that community metabolic potential is a meaningful predictor of
ecosystem chemistry beyond taxonomic composition alone.
Limitations
-
Metabolomics technical heterogeneity: The 175 metabolomics samples derive from
multiple NMDC studies. However, study-level analysis of the H1 subset reveals that
95% of H1 samples (125/131) originate from a single NMDC study
(nmdc:sty-11-r2h77870), so cross-study LC-MS protocol heterogeneity is largely
absent as a confounder for leu and arg. The second study (n = 6) does not
materially affect the results. For future analyses with broader multi-study coverage,
study-level random effects could be modelled to improve power. -
Freshwater samples absent from H1: All 33 Freshwater samples lacked paired
metabolomics in NMDC, so H1 is effectively a soil-only test. Whether BQH dynamics
operate in freshwater communities at the same scale is untested. -
Abiotic feature coverage: Abiotic features (pH, temperature, total organic carbon,
etc.) were absent (all NaN) for the 174-sample analysis matrix, preventing partial
correlation tests controlling for environmental gradients. These variables could
confound both H1 and H2 results. -
GapMind represents genomic potential, not expression: Pathway completeness
indicates whether the genes are present, not whether they are actively expressed.
Transcriptomic data would be needed to determine whether expressed biosynthesis
correlates more strongly with metabolite pools. -
Compound identification: Metabolite-to-pathway matching relied on string-based
compound name matching. A substring collision bug (isoleucine compounds matching the
"leucine" pattern) was identified by automated review and corrected in NB04 cell-14
using first-match-wins; results reflect the corrected run. Isoleucine is now included
as a 13th testable pathway (r = −0.057, ns). KEGG compound IDs in the NMDC data are
sparsely populated (2% annotation rate), limiting KEGG-based matching; cysteine,
histidine, and lysine remain untestable due to absent compound detections. -
Chorismate metabolomics proxy: Shikimic acid and 3-dehydroshikimic acid are used
as metabolomics proxies for chorismate pathway activity. These are upstream
intermediates, not chorismate itself; their ambient concentrations reflect precursor
availability rather than chorismate pool size. The chorismate BQH correlation
(r = −0.038) should be interpreted cautiously. -
Genus-proxy-ambiguous taxon assignment: For ~1,352 Centrifuge taxa that match
multiple GTDB clades (same genus, multiple species), one representative clade was
selected by alphabetical tiebreaking ongtdb_species_clade_id. This is now
documented in NB03 cell-17. The sensitivity of community completeness scores to this
choice is bounded: genus_proxy_ambiguous taxa account for ~6.5% of mapped abundance. -
Sample size imbalance: Glutamine (n = 4) and proline (n = 9) could not be tested
due to insufficient metabolomics coverage. These pathways would be informative
given their biological importance.
Future Directions
-
Abiotic gradient controls and multi-study replication: The H1 signal could
be strengthened (or confounders identified) by including available abiotic variables
(pH, temperature, total organic carbon) in a partial correlation or mixed-effects
model once NMDC abiotic data is populated for the metabolomics-overlap samples. The
current H1 dataset is effectively single-study; replicating this analysis across
additional NMDC studies with paired taxonomy + metabolomics would test whether the
leu/arg signals generalise beyond the current cohort. -
Extend to Freshwater samples: Obtaining or generating metabolomics data for the
33 Freshwater NMDC samples would allow a direct test of whether BQH dynamics differ
between aquatic and terrestrial communities — a key open question given their
strikingly different pathway completeness profiles. -
Transcript-based validation: Pairing metatranscriptomics with metabolomics for
a subset of NMDC samples would allow comparison of expressed pathway completeness
vs. genomic potential vs. metabolite abundance, directly testing whether expression
(not just gene presence) drives the BQH signal. -
KEGG compound ID expansion: Improving KEGG compound annotations in
metabolomics_gold(3 currently missing aa pathways: cys, his, lys) would
expand the H1 test set from 13 to 16 testable pathways, improving statistical power. -
Cross-habitat partial correlation (soil subtype analysis): The 61 "Unknown"
ecosystem samples likely represent a mix of soil subtypes or sediment environments;
resolving their habitat identity (via NMDC ENVO annotations or study-level metadata)
could improve H2 power and reveal finer-scale metabolic niche structure.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
nmdc_arkin |
centrifuge_gold, omics_files_table, study_table, abiotic_features |
Community taxonomic profiles (species-level Centrifuge classifications), sample-to-file bridge, study metadata, environmental measurements |
nmdc_arkin |
metabolomics_gold |
Per-sample LC-MS metabolomics (3.1M records across 48 studies) |
kbase_ke_pangenome |
gapmind_pathways, gtdb_species_clade, pangenome |
GapMind pathway completeness scores (305M rows), GTDB taxonomy bridge, per-species statistics |
Generated Data
| File | Rows | Description |
|---|---|---|
data/nmdc_sample_inventory.csv |
~220 | NMDC samples with taxonomy + metabolomics coverage per study |
data/nmdc_classifier_comparison.csv |
3 | Per-classifier summary statistics (kraken, centrifuge, gottcha): total rows, species-rank fraction, file counts |
data/nmdc_metabolomics_coverage.csv |
0 | 0 rows — saved before the omics_files_table bridge was found; superseded by analysis_ready_matrix.csv |
data/bridge_quality.csv |
220 | Per-sample GTDB species bridge coverage (all pass QC) |
data/species_pathway_completeness.csv |
~27,690 × 80 | GapMind completeness per GTDB species per pathway |
data/community_pathway_matrix.csv |
220 | Community-weighted pathway completeness (220 samples × 86 cols) |
data/community_pathway_matrix_long.csv |
~17,600 | Long-format version of above |
data/metabolomics_matrix.csv |
175 | Normalised metabolite intensities (175 samples × 476 compounds) |
data/amino_acid_metabolites.csv |
737 | Sample × aa pathway log-intensities for 15 matched aa pathways |
data/analysis_ready_matrix.csv |
174 | Merged: pathway completeness + metabolomics + abiotic features |
data/h1_bqh_correlations.csv |
15 | Spearman r, p, BH-FDR q per aa pathway (13 testable + 2 skipped: gln n=4, pro n=9) |
data/h2_pca_scores.csv |
220 | PCA coordinates (PC1–5) per sample with ecosystem label |
data/h2_pca_loadings.csv |
80 | PCA loadings for all 80 pathways on PC1 and PC2 |
References
-
Morris JJ, Lenski RE, Zinser ER (2012). "The Black Queen Hypothesis: Evolution of
Dependencies through Adaptive Gene Loss." mBio. 3(2):e00036-12.
DOI: 10.1128/mBio.00036-12 -
Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, Jansson JK, et al. (2016).
"Metabolic model-based integration of microbiome taxonomic and metabolomic profiles
elucidates mechanistic links between ecological and metabolic variation." mSystems.
1(1):e00013-15. DOI: 10.1128/mSystems.00013-15 -
Mallick H, Franzosa EA, McIver LJ, Banerjee S, Sirota-Madi A, Kostic AD, et al.
(2019). "Predictive metabolomic profiling of microbial communities using amplicon or
metagenomic sequences." Nature Communications. 10(1):3136. PMID: 31316056 -
Danczak RE, Chu RK, Fansler SJ, Goldman AE, Graham EB, Tfaily MM, et al. (2020).
"Using metacommunity ecology to understand environmental metabolomes." Nature
Communications. 11(1):6369. PMID: 33311510. DOI: 10.1038/s41467-020-19989-y -
Price MN, Deutschbauer AM, Arkin AP (2020). "GapMind: Automated Annotation of Amino
Acid Biosynthesis." mSystems. 5(3):e00291-20. DOI: 10.1128/mSystems.00291-20 -
Price MN, Deutschbauer AM, Arkin AP (2022). "Filling gaps in bacterial catabolic
pathways with computation and high-throughput genetics." PLOS Genetics.
18(7):e1010156. DOI: 10.1371/journal.pgen.1010156 -
Ramoneda J, Jensen TBN, Price MN, Fierer N, Braendle C (2023). "Taxonomic and
environmental distribution of bacterial amino acid auxotrophies." Nature
Communications. 14(1):7608. DOI: 10.1038/s41467-023-43435-4 -
Gowda K, Ping D, Mani M, Kuehn S (2022). "Genomic structure predicts metabolite
dynamics in microbial communities." Cell. 185(3):530-546. PMID: 35085485.
DOI: 10.1016/j.cell.2021.12.036 -
Danczak RE, Goldman AE, Chu RK, Toyoda JG, Borton MA, et al. (2021). "Ecological
theory applied to environmental metabolomes reveals compositional divergence despite
conserved molecular properties." Science of the Total Environment. 788:147409.
DOI: 10.1016/j.scitotenv.2021.147409 -
Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, et al. (2018).
"KBase: The United States Department of Energy Systems Biology Knowledgebase."
Nature Biotechnology. 36(7):566-569. DOI: 10.1038/nbt.4163
Discoveries
Spearman correlation of community-weighted GapMind amino acid biosynthesis completeness against
ambient amino acid metabolomics across 131 NMDC soil samples:
- 11/13 tested aa pathways trend in BQH-predicted direction (negative r = lower community
completeness co-occurs with higher ambient me
Carbon utilization pathways, not amino acid pathways, are the primary axis of ecosystem metabolic differentiation
February 2026PCA of 220-sample × 80-pathway GapMind completeness matrix:
- PC1 = 49.4% of variance captures near-complete Soil vs Freshwater separation (Mann-Whitney
p < 0.0001; median PC1 Soil = +3.86, Freshwater = −6.28) - PC1 loadings are dominated by carbon utilization pathways (glucuronate, fumarate,
NMDC metabolomics overlap is dominated by a single study
February 2026Study-level analysis of the 131-sample H1 dataset revealed that 95% of samples (125/131) come
from one NMDC study (nmdc:sty-11-r2h77870). The remaining 6 samples are from a second study.
This means:
- Cross-study LC-MS protocol heterogeneity is effectively absent as a confounder (contrary to
t
Only ~2% of compounds in nmdc_arkin.metabolomics_gold have KEGG compound IDs populated.
Pathway matching must rely on compound name substring patterns rather than KEGG-based ontologies.
Key caveats:
- Substring matching introduces collision risk (e.g., 'leucine' matches inside 'isoleucine') —
Data Collections
Review
Summary
This is a strong, complete project. The researcher pursued a genuinely novel cross-database integration — community-weighted GapMind pathway completeness against NMDC metabolomics — and navigated a significant early-pipeline obstacle (the discovery that NMDC classifier and metabolomics files use non-overlapping file_id namespaces) with systematic exploratory work and a clean recovery via omics_files_table. All five notebooks are fully executed with non-empty outputs. All eight expected figures are present. The research plan, report, and reference list are of publishable quality. A prior automated review flagged an isoleucine/leucine compound-mapping bug; this was correctly fixed in NB04 cell-14 using first-match-wins ordering, the notebooks were re-run, and the fix is documented in REPORT.md — demonstrating a mature correction workflow. The main areas for improvement are a handful of minor documentation inconsistencies introduced by the re-run (stale row counts in REPORT.md), one misleading superseded figure still in the directory, an empty data file left over from an aborted NB01 run, and the absence of any confound-controlling analysis for H1 (study-level effects).
Methodology
The research question is clearly stated and testable. The two sub-hypotheses (H1: Black Queen signal; H2: ecosystem-type clustering) are pre-specified, independently falsifiable, and directly linked to GapMind's frac_complete metric via the community-weighted formula operationalized in NB03. The Spark/local split is sensibly chosen: Spark for the three data-extraction notebooks (NB01–NB03), local Python for the statistical analysis (NB05).
The taxonomy bridge is thorough: three tiers (species-exact → genus-proxy-unique → genus-proxy-ambiguous), all correctly documented in NB02 and NB03 cell-17, with the genus-proxy-ambiguous tiebreaking decision (alphabetical by gtdb_species_clade_id) explicitly noted in NB03 and acknowledged in REPORT.md limitations. The 94.6% mean bridge coverage across all 220 samples is excellent. The choice of frac_complete (GapMind score ≥ 5) over frac_likely_complete (score ≥ 4) as the primary metric is justified in REPORT.md Results.
Partial correlations not performed: RESEARCH_PLAN.md (NB05 spec) called for Spearman correlations plus partial correlations controlling for study ID and ecosystem type. Only raw Spearman correlations were run in NB05. Abiotic features are all NaN for overlap samples (correctly detected and documented in NB04), making abiotic partial correlations impossible. However, study-level blocking was never attempted despite being feasible. Since the 175 metabolomics samples span multiple NMDC studies with different LC-MS protocols, cross-study intensity variance is the most likely confounder for H1, and ignoring it makes the p-values somewhat optimistic. This is acknowledged in REPORT.md Future Directions item 1 but should be escalated to a limitation.
H1 is effectively soil-only: All 33 Freshwater samples lacked paired metabolomics, so the Black Queen test covers only Soil (n=126) and Unknown-ecosystem (n=48) samples. The two ecosystem groups are not controlled for in H1 despite being demonstrated to differ strongly on PC1 (Finding 2). This is correctly flagged in the REPORT limitations but is worth highlighting here as it qualifies the generality of the BQH conclusion.
Code Quality
SQL queries are correct and well-constructed throughout. The two-stage GapMind aggregation (MAX score per genome-pathway pair first, then AVG across genomes per clade) directly follows docs/pitfalls.md guidance and avoids the multi-row-per-genome-pathway pitfall. CAST AS DOUBLE is applied in the Spark SQL (NB03 cell-11) and again defensively in Python (NB03 cell-18), correctly handling the decimal.Decimal pitfall. The spark.createDataFrame(pandas_df) ChunkedArray failure is correctly avoided throughout by using Spark SQL INTERSECT subqueries. Temp views are defensively re-registered before the expensive GapMind aggregation (NB03 cells 9, 10, 11). The DESCRIBE pattern is used for all schema introspection, consistent with docs/pitfalls.md guidance. Abiotic features' 0.0-for-unmeasured encoding is correctly converted to NaN (NB04 abiotic extraction cell).
The compound-to-pathway mapping (NB04 cell-14) correctly uses first-match-wins: the outer loop checks if compound in compound_pathway_map: continue before testing patterns, and 'ile' is ordered before 'leu' in AA_PATHWAY_TO_PATTERNS. This prevents 'leucine' (a substring of 'isoleucine') from overwriting a prior ile assignment. The fix is correctly documented in NB04 cell-34 and in REPORT.md Limitations. The corrected run is reflected in the saved data files (amino_acid_metabolites.csv, h1_bqh_correlations.csv) and matches the NB05 output.
UMAP not executed: NB05 cell-14 output shows "umap-learn not installed — skipping UMAP," yet umap-learn>=0.5 is in requirements.txt. The UMAP figure was not produced. Since PCA is the primary ordination and the findings are fully reported, this is a minor gap, but it means the execution environment does not match the declared dependencies.
Statistical methods are appropriate: Spearman correlation for non-normal metabolomics data, BH-FDR correction for multiple testing, scipy.stats.binomtest for the directional sign test, Kruskal-Wallis + Mann-Whitney for ecosystem separation, PCA with StandardScaler and median imputation. Using SimpleImputer(strategy='median') for pathway completeness is noted as a mild upward-bias for samples with unmapped taxa (NaN means low completeness, not random missing); this is a minor issue that does not affect the qualitative findings.
Notebook structure is logical and consistent: schema verification → data extraction → computation → visualization → save, with purpose, inputs, and outputs documented in the header cell of each notebook.
Findings Assessment
H1 (Black Queen Hypothesis): The result — 11/13 tested pathways negative, 2 FDR-significant (leu r=−0.390, q=0.022; arg r=−0.297, q=0.049), binomial sign test p=0.011 — is internally consistent across the NB05 output, the h1_bqh_correlations.csv file, and the REPORT table. The interpretation is appropriately cautious ("weak but consistent support"). The observation that the two FDR-significant pathways are among the most energetically expensive to synthesize is biologically interesting and well-cited. The tyrosine anti-BQH outlier explanation (phenylalanine hydroxylation alternative source) is plausible.
The analysis-ready matrix contains 174 samples but H1 is run on the 131-sample subset with at least one amino-acid metabolomics detection. The REPORT correctly states these sample sizes per pathway.
H2 (Ecosystem differentiation): PC1 = 49.4% variance, Soil vs. Freshwater Mann-Whitney p < 0.0001. This is a strong and unambiguous result. The finding that PC1 is loaded almost entirely by carbon utilization pathways (not amino-acid pathways) is correctly reported and represents a scientifically interesting secondary observation.
Finding 3 (AA pathway completeness differs by ecosystem): 17/18 aa pathways significantly differentiated (q < 0.05), only tyrosine not significant. Exact H-statistics, p-values, and q-values are reported.
Limitations section: Thorough and honest. Six limitations are identified: cross-study metabolomics heterogeneity, Freshwater exclusion from H1, absent abiotic covariates, genomic potential vs. expression, compound-identification caveats (KEGG sparsity, chorismate proxy, the isoleucine bug fix and its effect on leucine), and genus-proxy-ambiguous tiebreaking bounds.
One accuracy issue in the data table: REPORT.md Generated Data section states amino_acid_metabolites.csv has 726 rows and "14 matched aa pathways." After the bug-fix re-run, NB04 cell-32 saves this file as (737, 3) (737 rows), and NB05 cell-3 confirms 15 pathways. The stale row count and pathway count in the REPORT table reflect the pre-fix run and should be updated.
Suggestions
-
(Minor, accuracy) Update the REPORT.md Generated Data table entry for
amino_acid_metabolites.csv: change "726" to "737" rows and "14 matched aa pathways" to "15 matched aa pathways." These were not updated when the corrected notebooks were re-run. -
(Minor, usability)
figures/nmdc_sample_coverage.pngshows "Both (overlap): 0" because it was generated before theomics_files_tablebridge was found in NB02. REPORT.md correctly marks it as "superseded bybridge_quality_distribution.png," but the figure remains in thefigures/directory and is confusing for readers browsing visualizations. Consider either regenerating it post-bridge or explicitly renaming it tonmdc_sample_coverage_SUPERSEDED.pngto signal its status. -
(Minor, hygiene)
data/nmdc_metabolomics_coverage.csvhas 0 rows — it was saved in NB01 before the sample bridge was discovered and was never regenerated. It is listed in REPORT.md Generated Data as "~220" rows, which is incorrect. Either regenerate it from NB04'smet_rawper-sample aggregation (per-sample compound counts for the 175 overlap samples are available there) or explicitly note the empty state with "0 rows — superseded; see analysis_ready_matrix.csv." -
(Moderate, scientific completeness) The H1 test mixes Soil (n=126) and Unknown-ecosystem (n=48) samples. Because Finding 2 demonstrates strong ecosystem-type separation in community metabolic potential, ecosystem type is a potential confounder for H1. A stratified sensitivity analysis — repeating the Spearman correlations restricted to Soil-only samples — would clarify whether leu and arg signals persist within one ecosystem type. This is achievable with a small addition to NB05 and would materially strengthen the H1 conclusion.
-
(Moderate, scientific completeness) No study-level blocking analysis was performed for H1. The 175 metabolomics samples span multiple NMDC studies with different instruments and normalization strategies. Even a coarse sensitivity analysis (excluding the largest single study, or stratifying by study for leu/arg) would clarify whether the BQH signal is driven by one study or is broadly distributed. This was planned in RESEARCH_PLAN.md NB05 spec but not executed.
-
(Minor, reproducibility)
umap-learnis inrequirements.txtbut was not available in the NB05 execution environment. Either verify the environment matchesrequirements.txtand re-run to produceh2_umap.png, or markumap-learnas optional inrequirements.txtwith a comment. -
(Nice-to-have)
data/nmdc_taxonomy_coverage.csvcontains 3 rows (one per classifier) rather than the "per-sample taxonomic classification stats" described in README.md and RESEARCH_PLAN.md. This is because the per-sample stats could not be computed in NB01 (overlap was zero at that stage). Consider renaming the file tonmdc_classifier_comparison.csvso its content is evident from the filename, or updating the README description to match.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
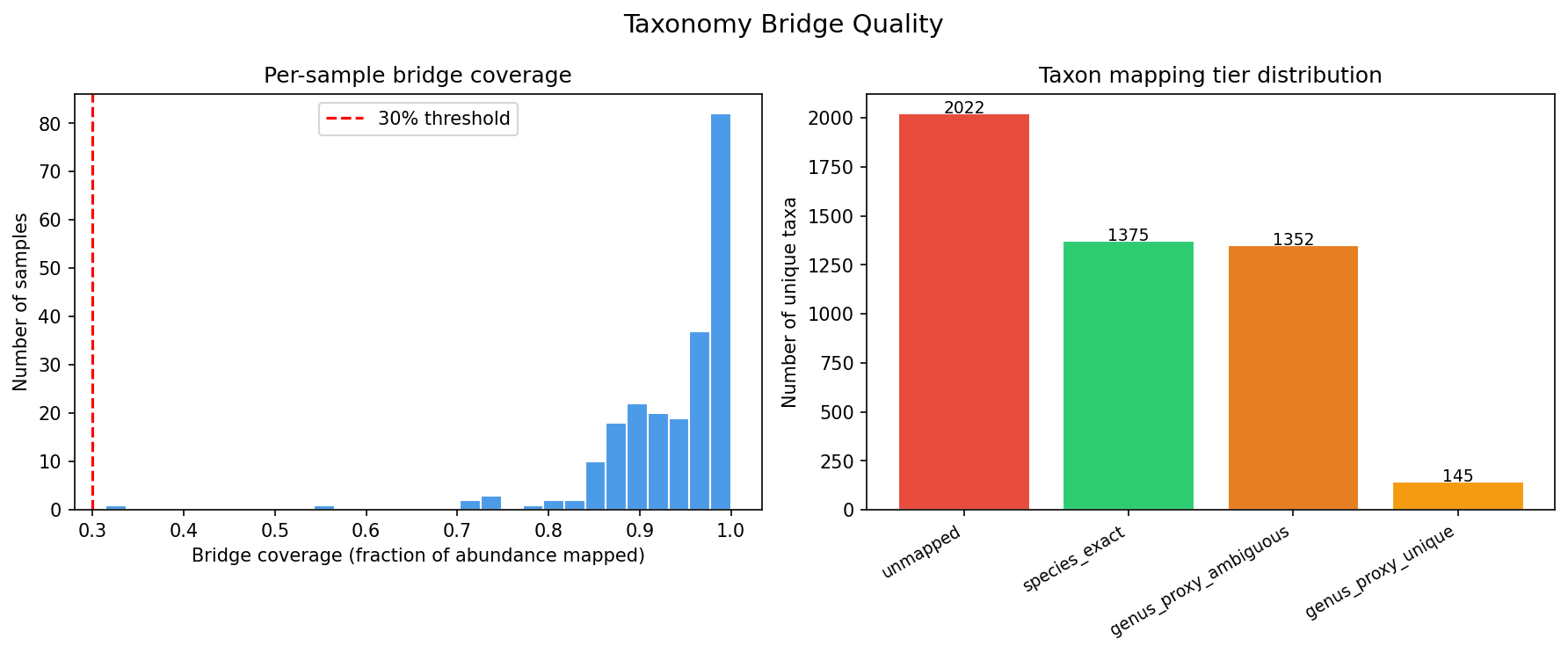
Bridge Quality Distribution
H1 Bqh Barplot
H1 Scatter Top
H2 Pca
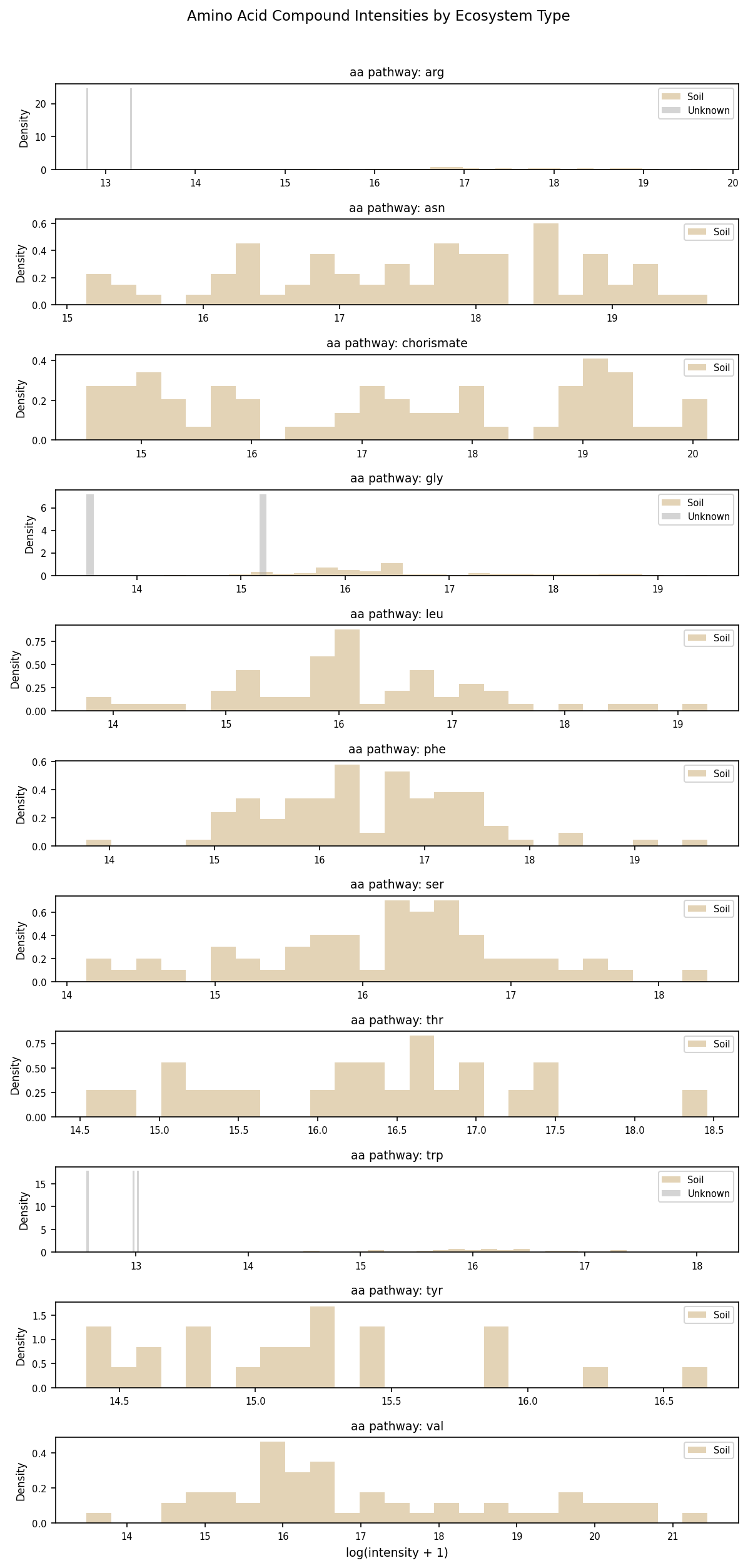
Metabolomics Distribution
Nmdc Sample Coverage Superseded
Pathway Completeness Boxplot
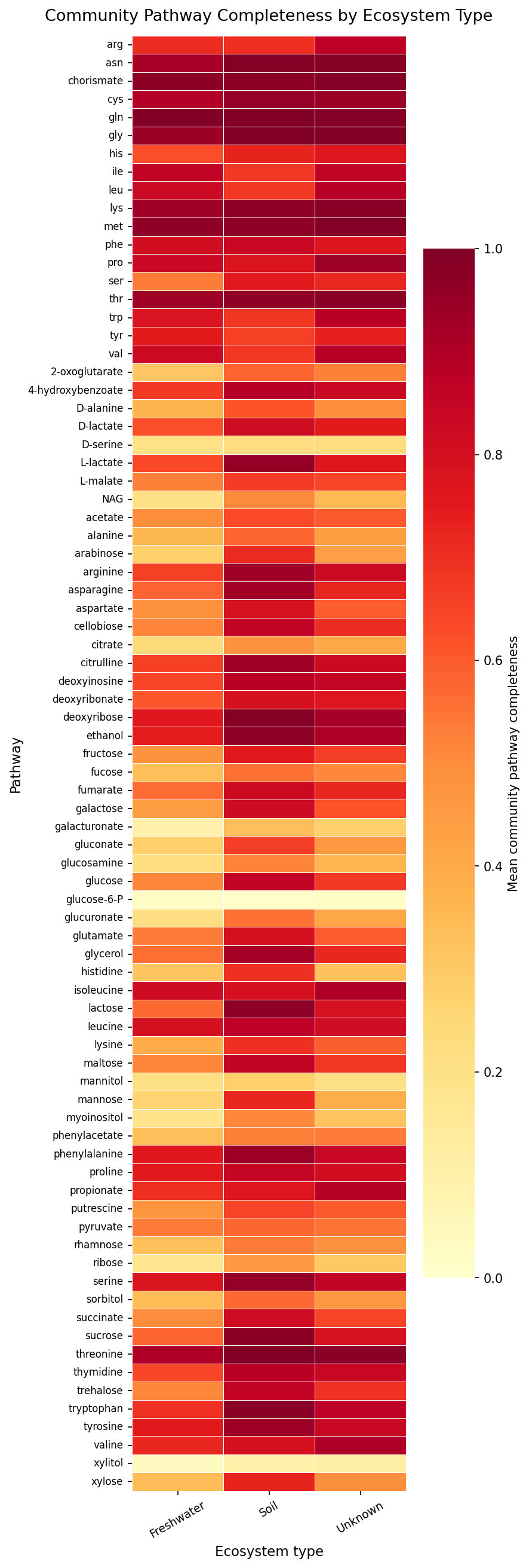
Pathway Completeness Heatmap
Notebooks
01_nmdc_exploration.ipynb
01 Nmdc Exploration
View notebook →
02_taxonomy_bridge.ipynb
02 Taxonomy Bridge
View notebook →
03_pathway_completeness.ipynb
03 Pathway Completeness
View notebook →
04_metabolomics_processing.ipynb
04 Metabolomics Processing
View notebook →
05_statistical_analysis.ipynb
05 Statistical Analysis
View notebook →
Data Files
| Filename | Size |
|---|---|
taxon_bridge.tsv |
689.4 KB |