Aromatic Catabolism Support Network in ADP1
CompletedResearch Question
Why does aromatic catabolism in Acinetobacter baylyi ADP1 require Complex I (NADH dehydrogenase), iron acquisition, and PQQ biosynthesis when growth on other carbon sources does not?
Research Plan
Hypothesis
- H0: The quinate-specific growth defects of Complex I, iron acquisition, and PQQ genes are unrelated to aromatic catabolism — they reflect general housekeeping requirements that happen to be masked on other carbon sources by the assay's limited dynamic range.
- H1: Aromatic catabolism via the β-ketoadipate pathway creates specific metabolic dependencies on (a) Complex I for NADH reoxidation, (b) iron acquisition for the Fe²⁺-dependent protocatechuate 3,4-dioxygenase, and (c) PQQ for the quinoprotein quinate dehydrogenase, forming a coherent support network not required by non-aromatic carbon sources.
Approach
Data Overview
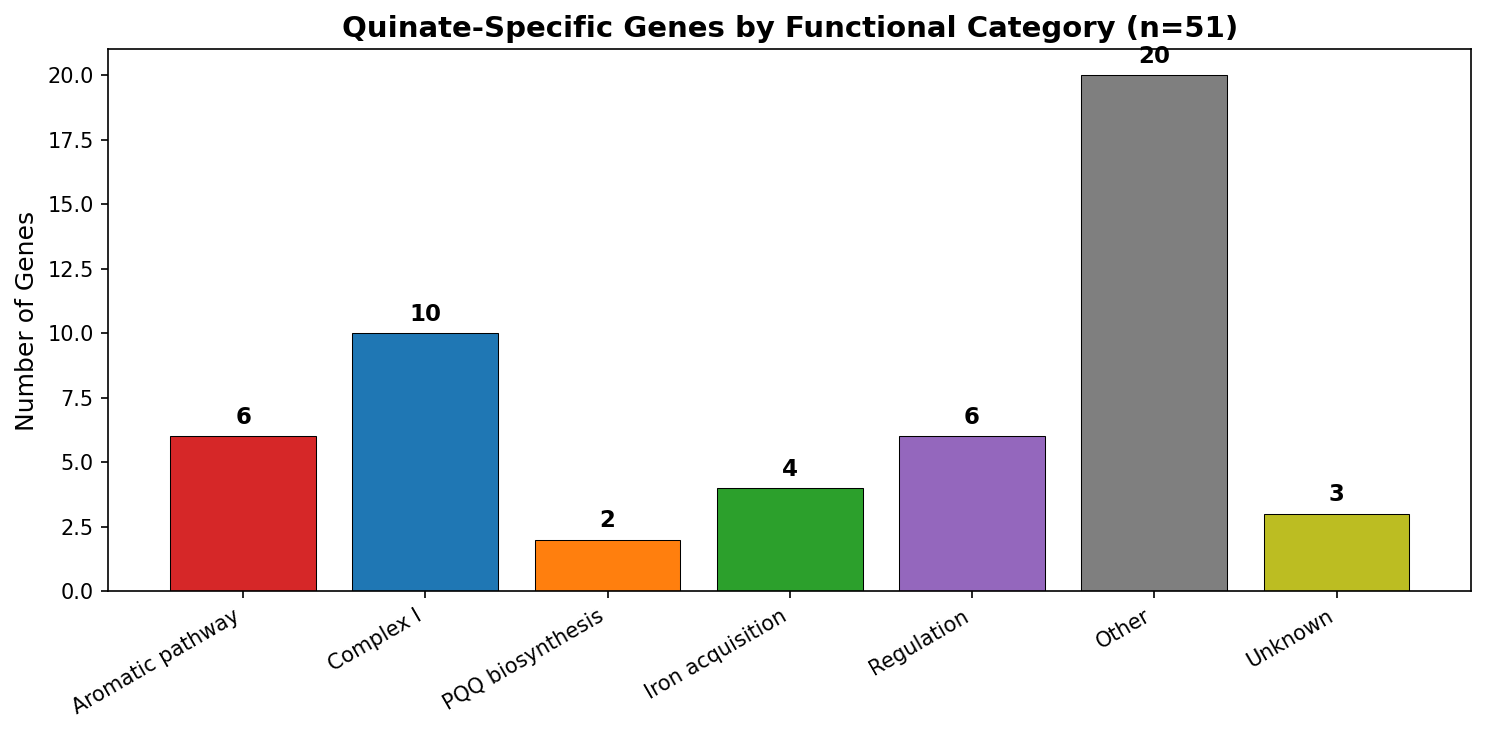
The analysis uses the 51 quinate-specific genes (specificity > 0.5, z-score < -1.0 on quinate) identified in projects/adp1_deletion_phenotypes/, categorized into functional groups:
| Category | Genes | Key members |
|---|---|---|
| Aromatic degradation pathway | 6 | pcaB, pcaC, pcaG, pcaH, quiA, quiB |
| Complex I (NADH dehydrogenase) | 10 | nuoA–N (chains A, E, F, G, H, I, J, K, M, N) |
| Iron acquisition | 3 | AcsD-like siderophore, ExbD/TolR, ferrichrome receptor |
| PQQ biosynthesis | 2 | pqqC, pqqD |
| Transcriptional regulation | 6 | LysR, Lrp, AraC, DeoR, IclR family regulators |
| Unknown/Other | 24 | Various — potential new pathway members |
Aim 1: Metabolic Dependency Mapping
Goal: Trace the biochemical logic connecting aromatic catabolism to each support system.
Methods:
- Map the quinate → TCA pathway step-by-step, annotating each reaction's cofactor requirements (Fe²⁺, PQQ, NAD⁺/NADH)
- Count NADH produced per mole of quinate catabolized vs glucose, acetate, and other carbon sources using the ADP1 FBA model (gene_reaction_data, genome_reactions tables)
- Compare FBA-predicted essentiality of the 51 genes across 9 aromatic vs 5 non-aromatic carbon sources using gene_phenotypes (230 conditions available)
- Test: does FBA predict the quinate-specificity of Complex I, or is this a gap in the model?
Aim 2: Operon and Regulon Structure
Goal: Determine whether the 51 quinate-specific genes are genomically clustered and co-regulated.
Methods:
- Plot gene positions along the ADP1 chromosome — are the support genes (Complex I, iron, PQQ) near the pca/qui cluster, or scattered?
- Identify operon structure from intergenic distances (<100 bp, same strand = likely same operon)
- Check whether the 6 transcriptional regulators control known aromatic degradation regulons
- Cross-reference with Dal et al. (2005) transcriptional organization
Aim 3: Cross-Species Conservation
Goal: Test whether the Complex I/aromatic catabolism dependency is conserved beyond ADP1.
Methods:
- Query kescience_fitnessbrowser for experiments where organisms were grown on aromatic substrates (benzoate, 4-hydroxybenzoate, protocatechuate, quinate). Check if any of the 48 FB organisms have aromatic-condition fitness data. Note: ADP1 is NOT one of the 48 FB organisms. Survey FB experiments in NB01 as a feasibility check.
- For organisms with data: do Complex I genes (KOs K00330–K00343) show condition-specific fitness defects on aromatics?
- Cross-species pangenome comparison: gene cluster IDs are species-specific and cannot be compared across species. Instead, use KO annotations via eggnog_mapper_annotations.KEGG_ko (joined on gene_cluster_id = query_name) to identify pca pathway orthologs (K00448, K00449, K01607, K01857, K01055, K05358, K03785) and Complex I orthologs (K00330–K00343) across Acinetobacter species. Test whether genomes carrying pca pathway KOs are more likely to also carry Complex I.
- Reference metal_fitness_atlas project for FB-to-pangenome organism mapping methodology
Aim 4: Characterize the "Other" Genes
Goal: Assign functions to the 24 quinate-specific genes without clear pathway assignments.
Methods:
- Build a co-fitness network using pairwise correlations across the 8-condition growth matrix: which of the 24 unknowns cluster with the pca pathway genes vs Complex I vs iron acquisition?
- Check if any of the 24 genes are in the same operons as known pathway genes
- Search for ortholog functions in other characterized aromatic-degrading bacteria
- Use the FBA model (gene_reaction_data) to check if any of the 24 genes map to reactions in the aromatic degradation subsystem
Revision History
- v2 (2026-02-19): Addressed plan reviewer feedback — specified KO-based cross-species comparison (gene clusters are species-specific), noted ADP1 not in FB, added FB feasibility check to NB01, referenced metal_fitness_atlas methodology
- v1 (2026-02-19): Initial plan
Overview
The prior project (adp1_deletion_phenotypes) identified 51 genes with quinate-specific growth defects. Unexpectedly, these include not just the 6 core aromatic degradation genes but also 10 Complex I subunits, 3 iron acquisition genes, 2 PQQ biosynthesis genes, and 6 transcriptional regulators. This project investigates whether these form a coherent metabolic dependency network — Complex I for NADH reoxidation during aromatic catabolism, iron for the Fe²⁺-dependent ring-cleavage dioxygenase, and PQQ for the quinoprotein quinate dehydrogenase — or whether the quinate-specificity is an artifact. Uses FBA predictions across 230 carbon sources (including 9 aromatics), genomic organization, co-fitness networks, and cross-species validation via the Fitness Browser.
Key Findings
1. Aromatic catabolism requires a 51-gene support network spanning 4 metabolic subsystems
The 51 quinate-specific genes in ADP1 organize into a coherent metabolic dependency network around the β-ketoadipate pathway. Co-fitness analysis assigns 44/51 genes (86%) to four functional subsystems: the core aromatic degradation pathway (8 genes), Complex I / NADH dehydrogenase (21 genes), iron acquisition (7 genes), and PQQ biosynthesis (2 genes), plus 6 transcriptional regulators. Each subsystem addresses a specific biochemical requirement of aromatic catabolism.
| Subsystem | Genes | Biochemical rationale |
|---|---|---|
| Aromatic pathway | 8 | Core: quinate → protocatechuate → β-ketoadipate → TCA cycle |
| Complex I | 21 | NADH reoxidation: TCA cycle generates excess NADH from aromatic catabolism |
| Iron acquisition | 7 | Fe²⁺ supply for protocatechuate 3,4-dioxygenase (ring cleavage) |
| PQQ biosynthesis | 2 | PQQ cofactor for quinoprotein quinate dehydrogenase |
| Regulation | 6 | Transcriptional control of aromatic degradation operons |
| Unassigned | 7 | Low co-fitness with all subsystems |
(Notebook: 01_metabolic_dependencies.ipynb, 03_cofitness_network.ipynb)
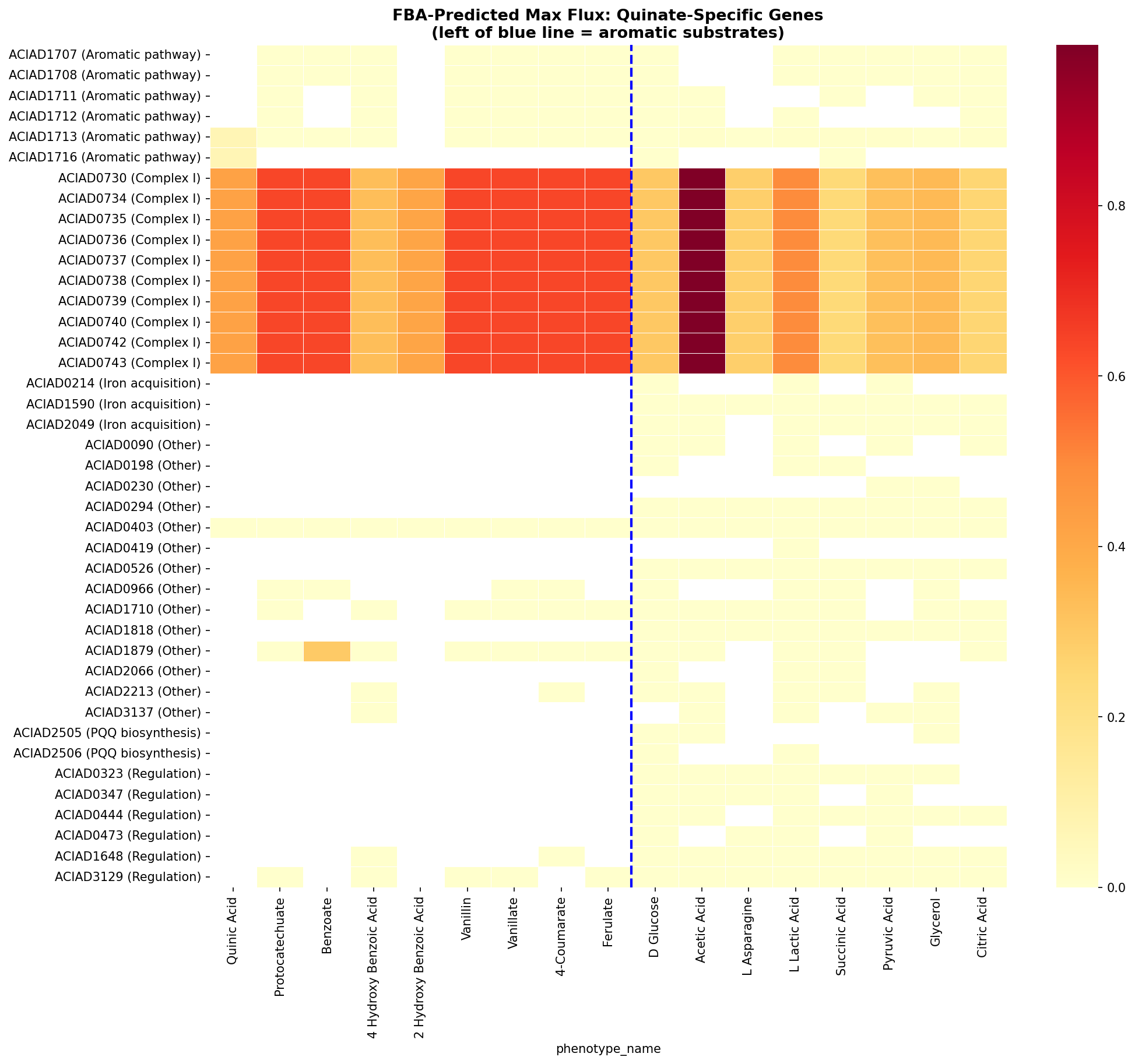
2. Complex I is the largest support subsystem — and invisible to FBA
Complex I (NADH:ubiquinone oxidoreductase) accounts for 21 of 51 quinate-specific genes (41%), making it the dominant support requirement for aromatic catabolism. The FBA model captures 1.76× higher Complex I flux on aromatic substrates (0.55 vs 0.31), but predicts 0% essentiality — it sees the increased demand but not the bottleneck. Furthermore, 30/51 quinate-specific genes have no FBA reaction mappings at all — cofactor supply chains (PQQ, iron) and regulatory genes are invisible to the metabolic model.
(Notebook: 01_metabolic_dependencies.ipynb)
3. Support subsystems are genomically independent but metabolically coupled
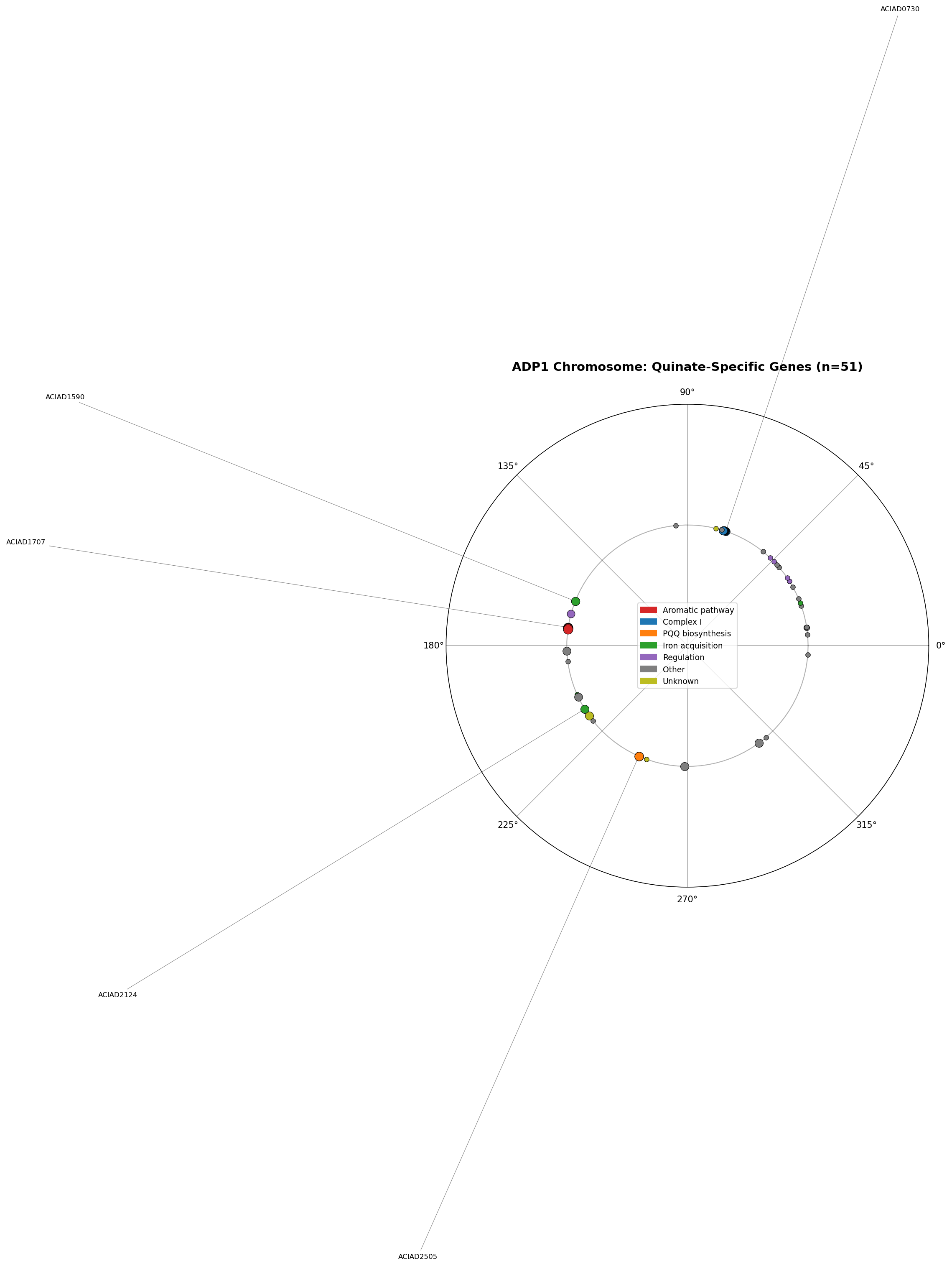
The four subsystems occupy distinct chromosomal locations: Complex I operon at 714–729 kb, pca/qui pathway at 1,709–1,724 kb, PQQ biosynthesis at 2,461 kb, and iron acquisition genes scattered across 4 loci. No cross-category operons exist (except one within the aromatic pathway itself). The metabolic dependency is not encoded by genomic co-localization — it emerges from the biochemistry of aromatic ring cleavage.
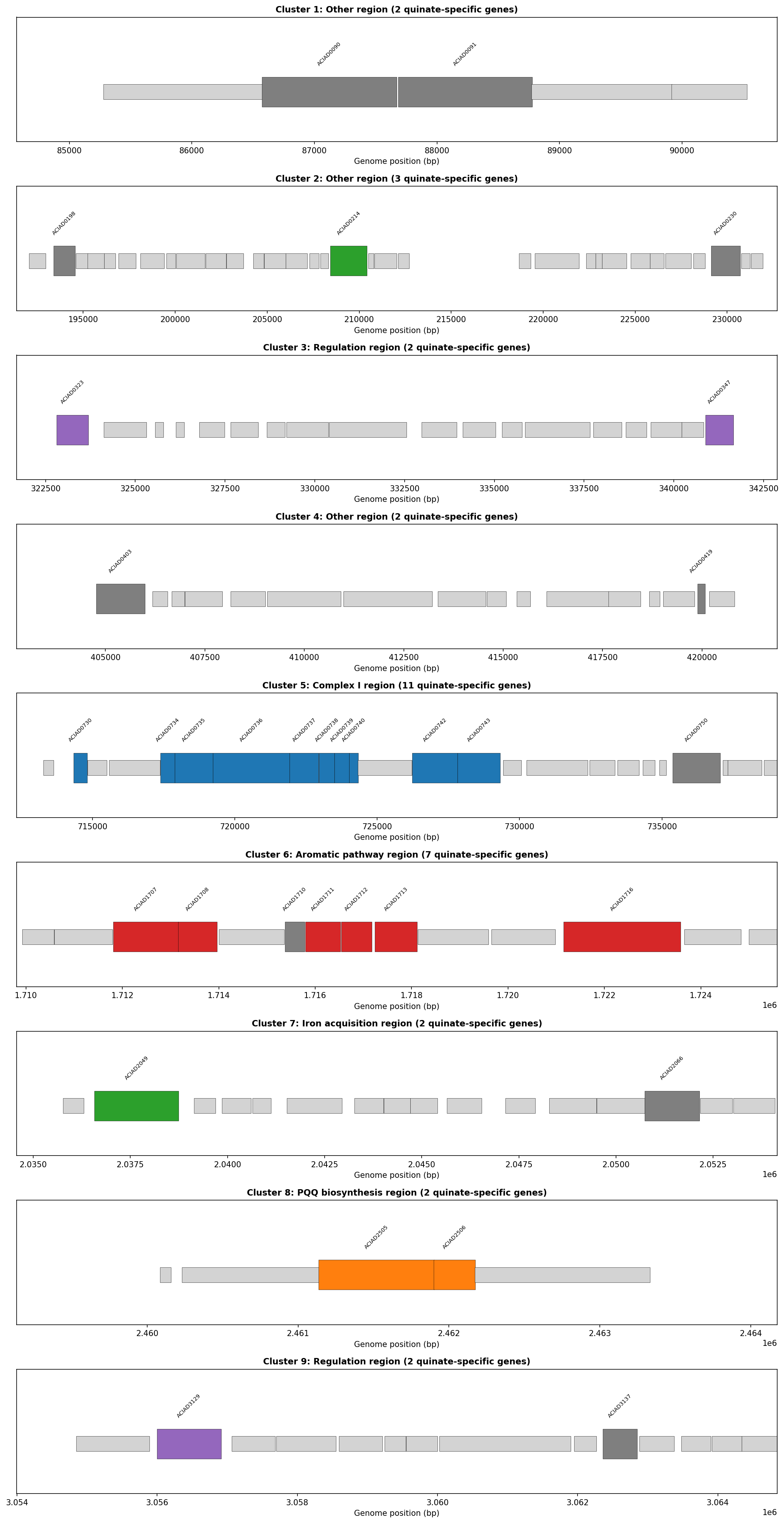
The Complex I operon contains 13 subunits (nuoA–N), all on the same strand with <100 bp intergenic distances. Disrupting any single subunit eliminates the entire complex, explaining why 10/13 subunits independently produce quinate-specific growth defects.
(Notebook: 02_genomic_organization.ipynb)
4. Co-fitness assigns 16 unknown genes to specific subsystems
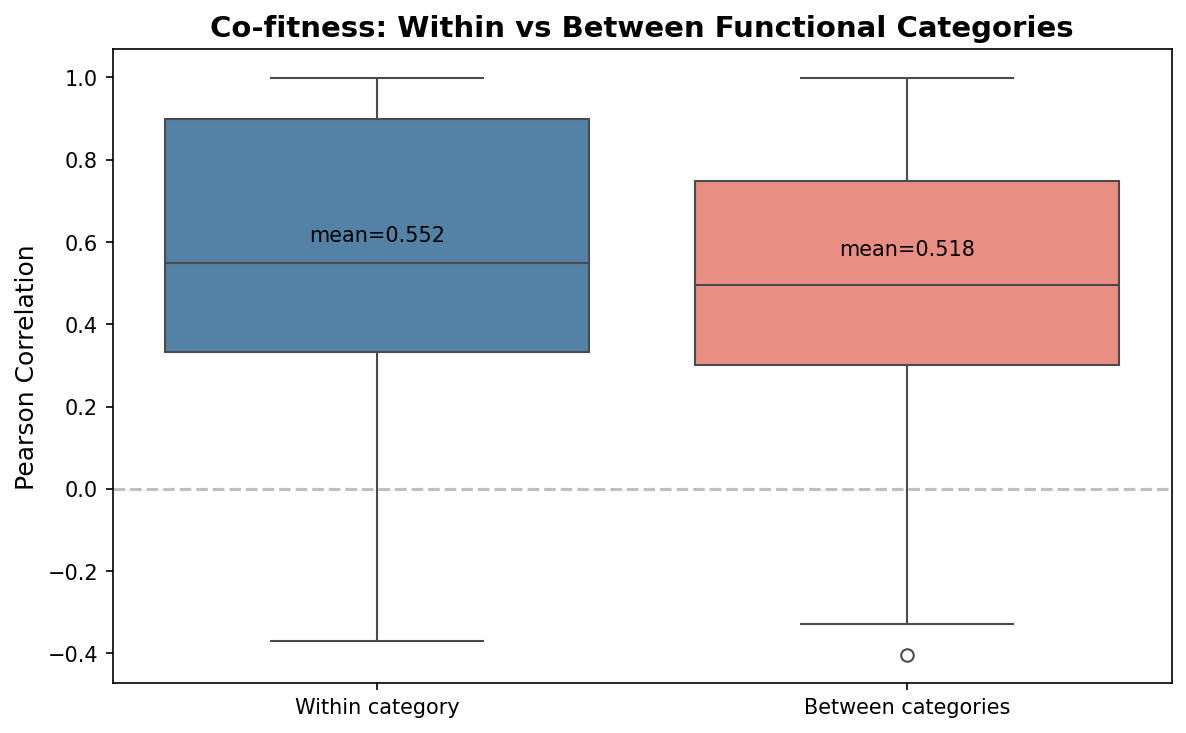
Of 23 genes initially categorized as "Other" or "Unknown," 16 were assigned to support subsystems with medium or high confidence based on growth profile correlations with known subsystem members. Two DUF-domain proteins (ACIAD3137, ACIAD2176) show r > 0.98 correlation with Complex I genes — these are candidates for uncharacterized Complex I accessory factors. Within-category correlation is dramatically higher than between-category (Complex I mean r = 0.992, Aromatic pathway r = 0.961).
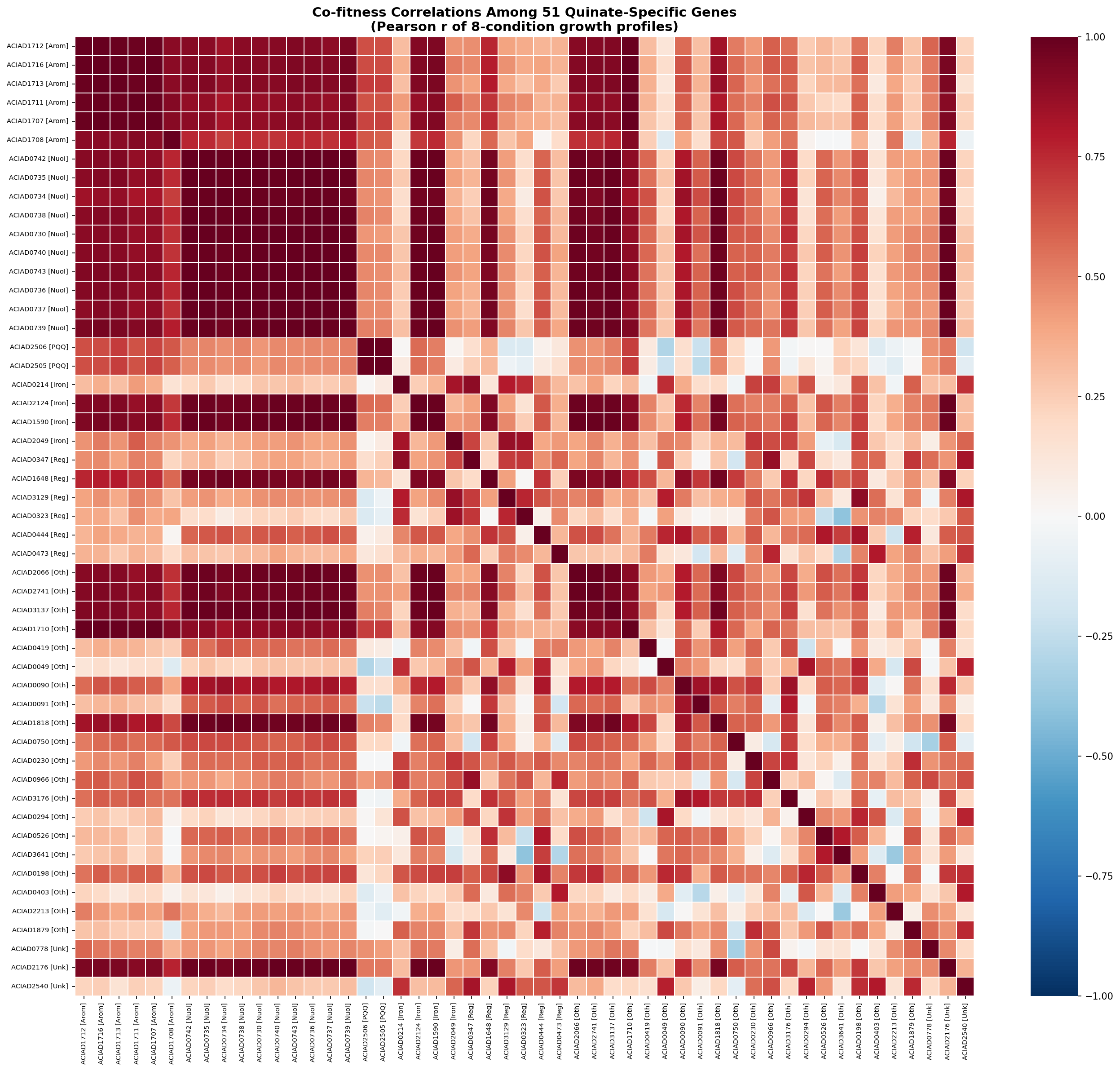
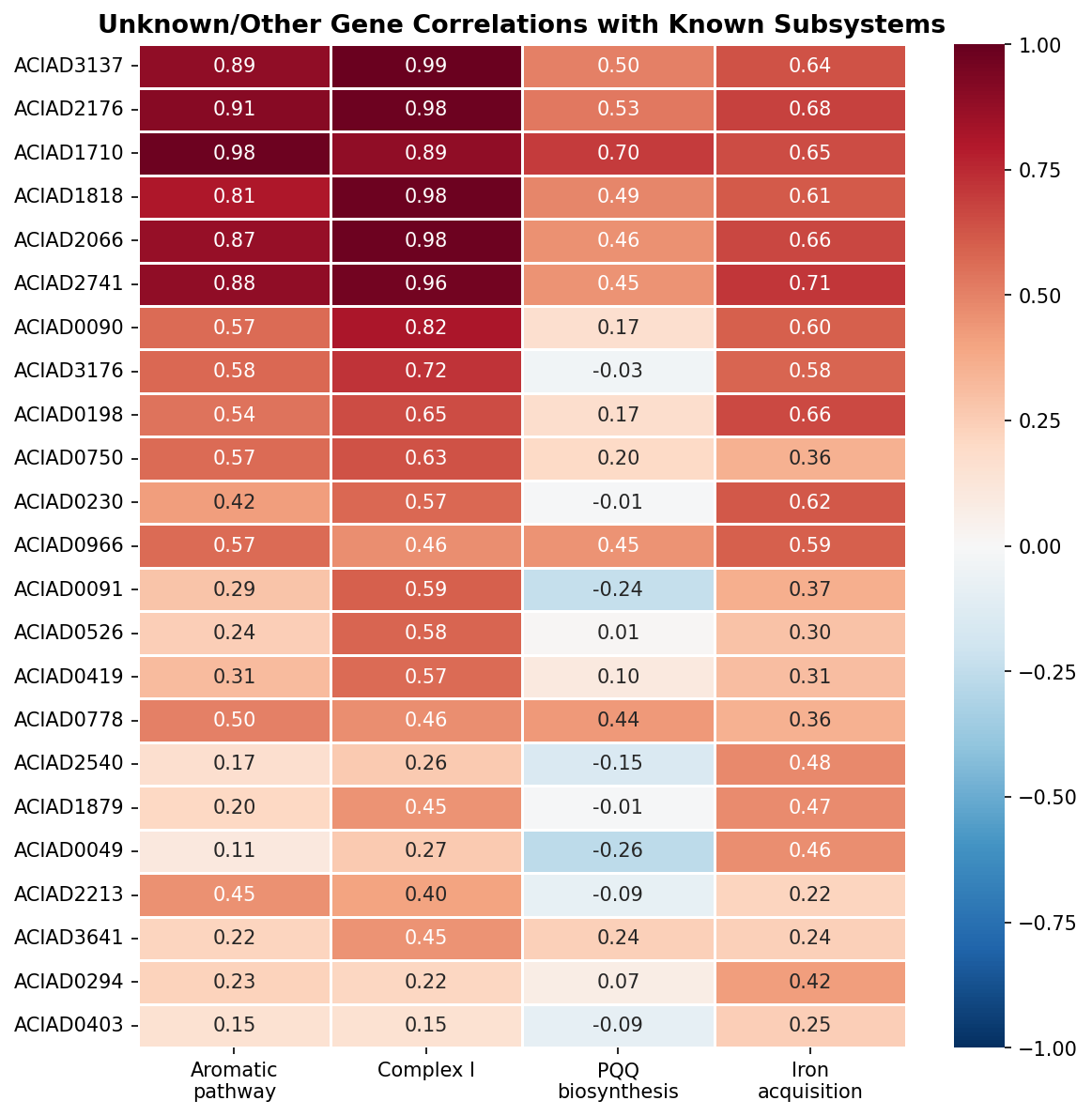
(Notebook: 03_cofitness_network.ipynb)
5. Cross-species: Complex I dependency is on high-NADH substrates, not aromatics specifically
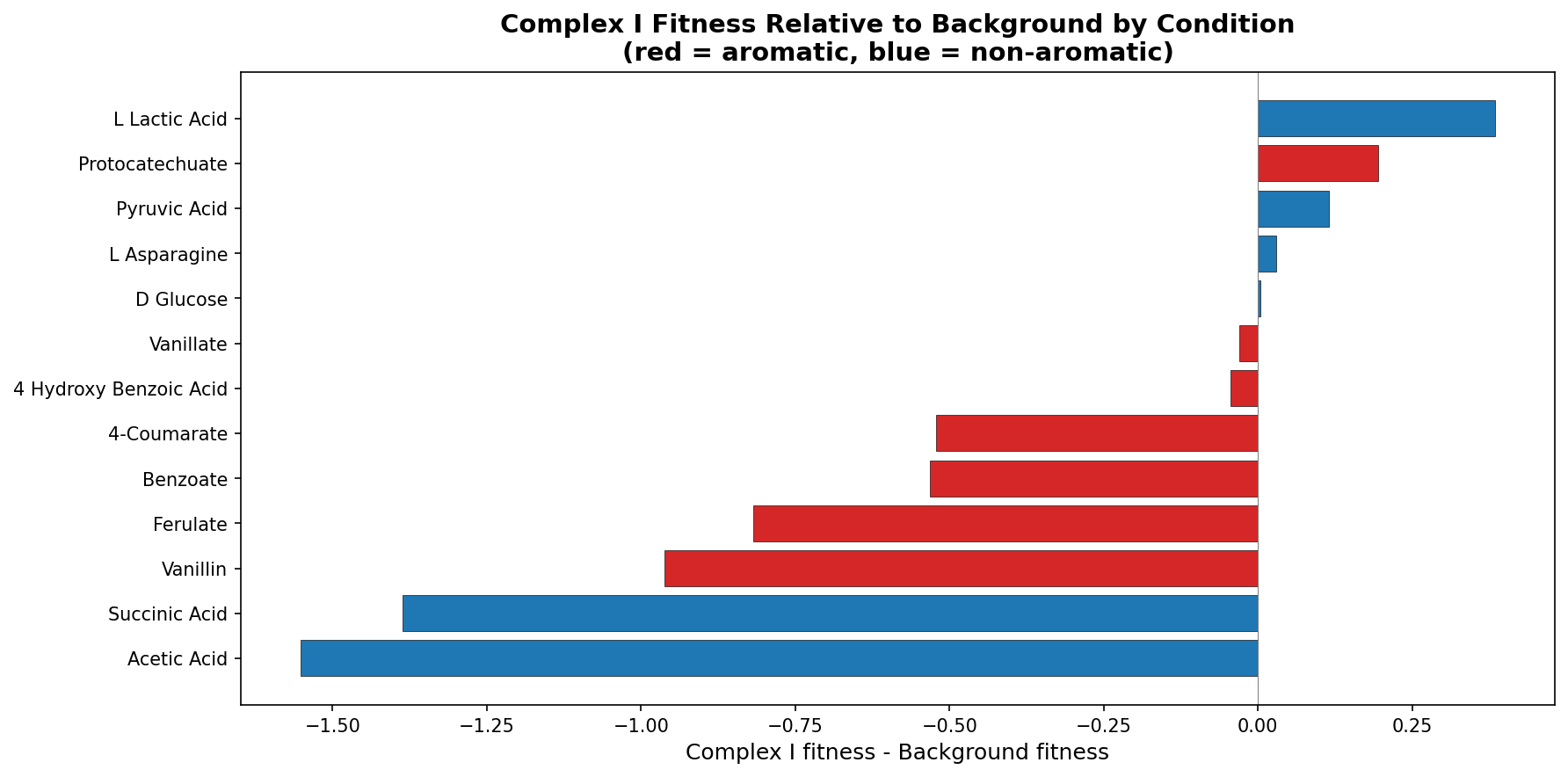
Ortholog-transferred fitness data from the Fitness Browser (12,241 entries, 2,005 genes, 13 conditions) shows Complex I orthologs have significantly worse fitness on aromatic conditions (mean = -1.35 vs -0.77, Mann-Whitney p < 0.0001). However, per-condition analysis reveals that the largest Complex I defects relative to background are on acetate (-1.55) and succinate (-1.39) — non-aromatic substrates that also generate high NADH flux through the TCA cycle. ADP1's quinate-specificity of Complex I likely reflects an alternative NADH dehydrogenase (NDH-2) that compensates on simpler substrates.
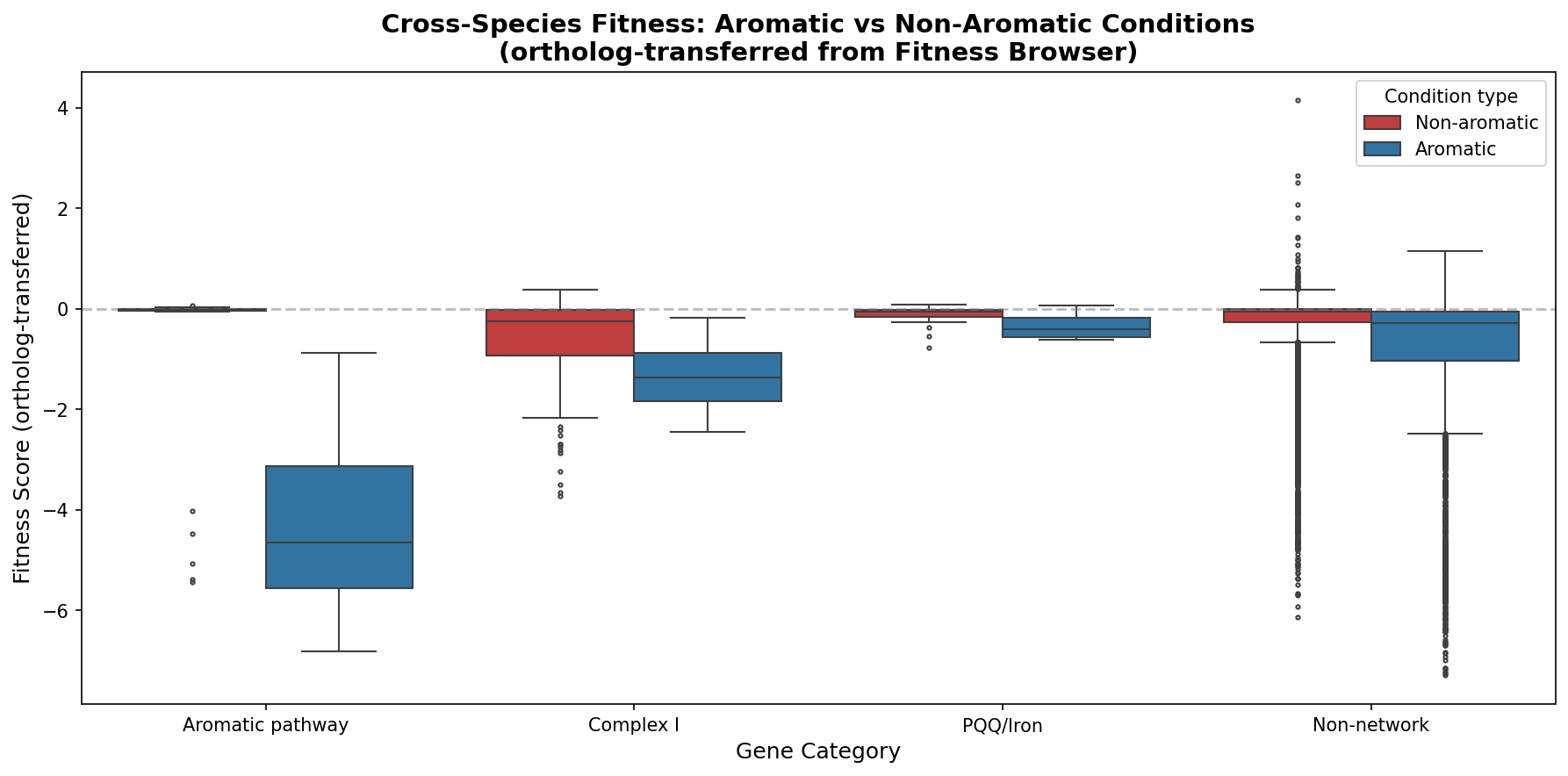
(Notebook: 04_cross_species.ipynb)
Results
The β-Ketoadipate Pathway and Its Cofactor Requirements
Quinate catabolism proceeds through protocatechuate to β-ketoadipate, ultimately yielding succinyl-CoA and acetyl-CoA that enter the TCA cycle. Three key enzyme steps create the support dependencies:
- Quinate dehydrogenase (quiA): A PQQ-dependent quinoprotein — requires PQQ cofactor, explaining the 2 PQQ biosynthesis genes (pqqC, pqqD)
- Protocatechuate 3,4-dioxygenase (pcaGH): A non-heme Fe²⁺-dependent ring-cleavage enzyme — requires iron, explaining the 7 iron acquisition genes (siderophore biosynthesis, ExbD/TolR transport, ferrichrome receptor, TonB-dependent receptor)
- TCA cycle oxidation: Succinyl-CoA and acetyl-CoA from β-ketoadipate are fully oxidized in the TCA cycle, generating NADH at multiple steps — requires Complex I for NADH reoxidation under high flux
FBA Model Blind Spots
The FBA model correctly predicts higher Complex I flux on aromatic substrates (1.76× ratio) but fails to identify it as a bottleneck (0% predicted essentiality). This is because FBA models optimize for growth rate and can redistribute flux through alternative pathways. In reality, Complex I operates as a single multi-subunit complex — loss of any subunit eliminates function entirely, creating a threshold effect that FBA's linear programming framework doesn't capture.
More fundamentally, 30/51 quinate-specific genes lack FBA reaction mappings. PQQ biosynthesis, iron acquisition, transcriptional regulation, and the newly identified Complex I accessory factors are outside the model's scope. This represents a systematic blind spot: the metabolic model captures core metabolism but not the cofactor supply chains and regulatory infrastructure that make it work.
Genomic Architecture
The support network's genomic organization reveals a separation between genetic and metabolic coupling:
- Within operons: genes are tightly co-regulated (Complex I: 13-gene operon, pca/qui: 12-gene operon)
- Between operons: no genomic linkage. Complex I, PQQ, and iron genes are on different parts of the chromosome
- 9 genomic clusters of ≥2 quinate-specific genes, with mild overall clustering (observed/expected NN distance ratio = 0.89)
Co-Fitness Network Validation
The co-fitness approach validates the functional categorization: Complex I genes correlate at r = 0.992 (essentially identical phenotypic profiles, consistent with single-operon co-regulation), and aromatic pathway genes at r = 0.961. The method successfully recovered pcaC (4-carboxymuconolactone decarboxylase), which was initially miscategorized by keyword matching, and identified two DUF proteins as probable Complex I accessory factors.
Interpretation
Literature Context
-
The PQQ and iron dependencies are well-established biochemistry: PQQ-dependent quinate dehydrogenases are documented across aromatic-degrading bacteria (Adachi et al. 2003), and protocatechuate 3,4-dioxygenase's non-heme Fe²⁺ requirement is a defining feature of intradiol dioxygenases (Que 2005). Our analysis quantifies these dependencies at the whole-genome level for the first time.
-
Stuani et al. (2014) showed 4/5 PQQ biosynthesis genes are upregulated on quinate vs succinate in ADP1. Our finding that PQQ genes are quinate-specific in growth phenotype is consistent with and extends their transcriptomic observation to the functional level.
-
The Complex I dependency is the novel finding. Bacteria typically have branched respiratory chains with both proton-pumping Complex I (NDH-1) and non-pumping NDH-2 (Melo & Teixeira 2016). NDH-2 can substitute for Complex I under low NADH flux but cannot match Complex I's capacity under high flux. Our cross-species data supports this: Complex I is dispensable on glucose and lactate (where NDH-2 suffices) but essential on substrates that generate high TCA cycle flux (aromatics, acetate, succinate).
-
Dal et al. (2005) mapped the transcriptional organization of the pca/qui genes. Our operon predictions match their experimental data, with the 12-gene pca/qui operon spanning pcaIJFBDCHG-quiABC plus transport genes.
Novel Contribution
-
Quantification of the support network: Previous work focused on individual pathway genes. This analysis reveals that aromatic catabolism requires 51 genes — over 8× the core pathway — organized into 4 biochemically rational subsystems. The support requirement exceeds the pathway itself by 7:1.
-
Complex I as the dominant support requirement: 21/51 quinate-specific genes (41%) are associated with Complex I, making NADH reoxidation the largest metabolic dependency of aromatic catabolism. This was not predicted by the FBA model and has not been systematically documented.
-
FBA model blind spot characterization: 30/51 genes have no FBA reaction mappings. This quantifies a specific class of metabolic model incompleteness — cofactor supply chains, iron homeostasis, and respiratory chain capacity constraints.
-
Co-fitness-based functional assignment: 16 previously uncharacterized genes assigned to specific subsystems, including two DUF proteins (ACIAD3137, ACIAD2176) as candidate Complex I accessory factors with r > 0.98.
-
Cross-species resolution: The Complex I dependency is not exclusively aromatic — it's a high-NADH-flux dependency that manifests on aromatics, acetate, and succinate. ADP1's apparent quinate-specificity reflects its specific respiratory chain architecture (NDH-2 compensating on simpler substrates).
Limitations
- The co-fitness analysis uses only 8 conditions (8-dimensional growth vectors), limiting the resolution of gene-gene correlations. With more conditions, the co-fitness network would provide sharper subsystem boundaries.
- Ortholog-transferred fitness data mixes signals from multiple FB organisms with different respiratory chain architectures. Direct Complex I fitness measurements on aromatic substrates in a single organism would be more definitive.
- The categorization of "Complex I-associated" genes beyond the core nuo operon (the 11 co-fitness assignments) is based on phenotypic correlation only. Some may have indirect connections rather than physical association with Complex I.
- PQQ biosynthesis genes also appear as glucose-specific in the adp1_deletion_phenotypes project (PQQ-dependent glucose dehydrogenase), so the PQQ dependency is not exclusively aromatic — it's shared with glucose catabolism.
Future Directions
-
Identify the alternative NADH dehydrogenase in ADP1: Search the ADP1 genome for NDH-2 (type 2 NADH dehydrogenase) genes. If present, their deletion phenotype on quinate vs glucose would directly test whether NDH-2 compensates for Complex I on non-aromatic substrates.
-
Validate DUF protein assignments experimentally: ACIAD3137 (UPF0234) and ACIAD2176 (DUF2280) show r > 0.98 co-fitness with Complex I. Protein-protein interaction studies or co-purification with Complex I would test whether these are physical accessory factors or indirect regulatory connections.
-
Expand the condition panel: The 8-condition growth matrix provides ~5 independent dimensions. Additional aromatic substrates (benzoate, catechol, vanillate) plus stress conditions (iron limitation, respiratory inhibitors) would refine the subsystem boundaries and test the NADH-flux hypothesis directly.
-
Cross-species pangenome comparison: Use BERDL's
eggnog_mapper_annotationsto test whether Acinetobacter species that carry the pca pathway are more likely to retain Complex I (KOs K00330–K00343) than species without aromatic degradation capability. -
Integrate with the metabolic model: The 30 genes without FBA reaction mappings represent a specific class of model incompleteness. Adding PQQ biosynthesis, iron homeostasis, and respiratory chain capacity constraints to the ADP1 FBA model could improve its predictive accuracy for condition-dependent essentiality.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
User-provided SQLite (berdl_tables.db) |
genome_features, gene_phenotypes, gene_reaction_data |
Growth ratios, FBA predictions (230 conditions), gene-reaction mappings |
kescience_fitnessbrowser |
Ortholog-transferred fitness via gene_phenotypes |
Cross-species fitness on aromatic substrates |
kbase_ke_pangenome |
Pangenome core/accessory annotations (via genome_features) | Conservation context |
Generated Data
| File | Rows | Description |
|---|---|---|
data/quinate_specific_genes.csv |
51 | Quinate-specific genes with growth profiles and annotations |
data/support_network_genes.csv |
51 | Genes with functional category assignments and biochemical rationale |
data/operon_assignments.csv |
51 | Operon membership and intergenic distances |
data/cofitness_network.csv |
1,275 | Pairwise Pearson correlations among 51 genes |
data/unknown_assignments.csv |
23 | Co-fitness-based subsystem assignments for Other/Unknown genes |
data/final_network_model.csv |
51 | Final category assignments integrating annotation + co-fitness |
data/cross_species_fitness.csv |
13 | Per-condition Complex I vs background fitness comparison |
References
- Adachi O et al. (2003). "PQQ-dependent quinoprotein dehydrogenases in oxidative fermentation." Enzyme and Microbial Technology.
- Dal S et al. (2005). "Transcriptional organization of genes for protocatechuate and quinate degradation from Acinetobacter sp. strain ADP1." Applied and Environmental Microbiology 71(2):1025-1034.
- de Berardinis V et al. (2008). "A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1." Molecular Systems Biology 4:174. PMID: 18319726
- Erickson E et al. (2022). "Critical enzyme reactions in aromatic catabolism for microbial lignin conversion." Nature Catalysis 5:886-898.
- Fischer R et al. (2008). "Catabolite repression of aromatic compound degradation in Acinetobacter baylyi." Journal of Bacteriology 190(5):1759-1767.
- Melo AMP & Teixeira M (2016). "Supramolecular organization of bacterial aerobic respiratory chains." Biochimica et Biophysica Acta - Bioenergetics 1857(3):190-197.
- Price MN et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557:503-509.
- Que L Jr (2005). "Non-heme iron dioxygenases: structure and mechanism." Structure and Bonding 97:21-58.
- Stanier RY & Ornston LN (1973). "The β-ketoadipate pathway." Advances in Microbial Physiology 9:89-151.
- Stuani L et al. (2014). "Novel metabolic features in Acinetobacter baylyi ADP1 revealed by a multiomics approach." Metabolomics 10(6):1223-1238. PMID: 25374488
Discoveries
The β-ketoadipate pathway in ADP1 has 8 core genes, but 51 genes total show quinate-specific growth defects — a 7:1 support-to-pathway ratio. The support genes organize into 3 biochemically rational subsystems: Complex I (21 genes, NADH reoxidation), iron acquisition (7 genes, Fe²⁺ for ring-cleavage
Read more →Of the 51 quinate-specific genes, 30 (59%) have no FBA reaction mappings — PQQ biosynthesis, iron acquisition, transcriptional regulators, and respiratory chain components are invisible to the metabolic model. For Complex I specifically, FBA predicts 1.76× higher flux on aromatics but 0% essentialit
Read more →Cross-species ortholog-transferred fitness data shows Complex I defects are largest on acetate (-1.55 vs background) and succinate (-1.39), not aromatics. ADP1's quinate-specificity likely reflects an alternative NADH dehydrogenase (NDH-2) that compensates on lower-NADH-flux substrates. The "aromati
Read more →ACIAD3137 (UPF0234/YitK) and ACIAD2176 (DUF2280) correlate at r > 0.98 with Complex I genes across 8 growth conditions. Both lack FBA reaction mappings and have no assigned metabolic function. Their near-perfect co-fitness with the nuo operon suggests physical or regulatory association with Complex
Read more →Data Collections
Review
Summary
This is a strong project that systematically investigates why aromatic catabolism in Acinetobacter baylyi ADP1 requires a 51-gene support network spanning Complex I, iron acquisition, and PQQ biosynthesis beyond the 6 core pathway enzymes. The analysis progresses logically through four notebooks: FBA-based metabolic dependency mapping, genomic organization and operon prediction, co-fitness network analysis for functional assignment of unknown genes, and cross-species validation via ortholog-transferred fitness data. The project produces a clear, biologically coherent model with an important nuance from the cross-species work: the Complex I dependency is on high-NADH-flux substrates generally, not aromatics exclusively. Documentation is thorough across README, RESEARCH_PLAN, and REPORT. All notebooks have saved outputs, 9 figures and 7 data files are generated, and a requirements.txt is provided. The main areas for improvement are: the categorize_gene() function is duplicated across 3 notebooks instead of being imported from the existing utils.py (which already contains a corrected version), the interaction test for Complex I aromatic specificity relies on only 5 genes, and the co-fitness assignment confidence scheme deserves clearer justification given the limited 8-condition dimensionality.
Methodology
Research question and hypotheses: The research question is clearly stated and testable. The explicit null (H0: quinate-specific defects are artifacts) vs alternative (H1: coherent metabolic dependency network) hypothesis structure in RESEARCH_PLAN.md provides a rigorous framework. The identification of potential confounders (growth ratio non-linearity, quinate's unusually high mean growth ratio, general Complex I importance for aerobic respiration) is a notable strength.
Multi-evidence approach: The four-aim design is well-conceived. Each notebook contributes independently:
- NB01: Establishes biochemical logic via FBA predictions and identifies model blind spots
- NB02: Tests genomic co-localization (finds the subsystems are physically independent)
- NB03: Provides functional grouping via co-fitness and reassigns 16 unknown genes
- NB04: Tests generality across species and reveals that the dependency is NADH-flux-based, not exclusively aromatic
This convergent evidence structure is appropriate for the question.
Data sources: Clearly identified in README and RESEARCH_PLAN. The project depends on a local SQLite database (berdl_tables.db, 136 MB) from a prior project and on ortholog-transferred fitness scores. The dependency chain (adp1_deletion_phenotypes -> this project) is documented.
Concern -- keyword categorization is fragile and inconsistently fixed: The categorize_gene() function in NB01 (cell 1), NB02 (cell 1), and NB03 (cell 1) uses keyword matching on RAST function descriptions and misclassifies ACIAD1710 (4-carboxymuconolactone decarboxylase, pcaC) as "Other" because the keyword list includes "muconate" but not "muconolactone." A utils.py file exists in the notebooks directory with a corrected version that includes "muconolactone" in the keyword list, but none of the notebooks import from it -- they all contain older, incorrect copies. This creates an inconsistency: the shared utility has the fix, but the notebooks that actually run don't use it. The pitfall is documented in docs/pitfalls.md (lines 733-737), but the fix hasn't been propagated to the notebook code.
Scope reduction acknowledged: The RESEARCH_PLAN's Aim 3 described querying kbase_ke_pangenome.eggnog_mapper_annotations for cross-species pangenome comparison of pca pathway and Complex I KO co-occurrence. This was replaced with ortholog-transferred fitness analysis in NB04 and appropriately deferred to Future Direction #4 in REPORT.md.
Code Quality
Notebook organization: All four notebooks follow a consistent setup -> analysis -> visualization -> summary pattern with markdown section headers. Each notebook ends with a summary cell printing key metrics. The markdown annotations explaining the pathway biochemistry (NB01, cell 2) and the ortholog transfer methodology note (NB04, cell 3) are valuable.
Code duplication (critical): The categorize_gene() function is copied verbatim into NB01 (cell 1), NB02 (cell 1), and NB03 (cell 1). The notebooks/utils.py module already exists with a corrected version (includes "muconolactone" keyword) but is not imported by any notebook. This is a maintenance problem and actively causes the ACIAD1710 misclassification in the first three notebooks. NB04 avoids the issue by loading final categories from final_network_model.csv (which incorporates the co-fitness correction from NB03).
SQL correctness: The queries against the SQLite database are straightforward and correct. NB04 (cell 4) correctly uses CAST(fitness_avg AS FLOAT) for string-typed Fitness Browser columns, consistent with the pitfall documented in docs/pitfalls.md (lines 63-69). NB01's FBA queries use parameterized placeholders for gene ID lists, which is good practice.
Statistical methods:
- Pearson correlation for co-fitness (NB03): Appropriate for continuous growth profiles. The population-level z-scoring using all 2,034 genes (NB03, cell 2) is good practice -- it avoids inflating correlations that would result from z-scoring only the 51 quinate-specific genes.
- Mann-Whitney U test (NB04, cell 11): Correctly applied as a non-parametric test. However, the interpretation needs care: both Complex I AND background genes show significantly worse fitness on aromatics (both p<0.0001). The interaction test (cell 12) is the biologically meaningful comparison and relies on only n=5 Complex I genes with data on both aromatic and non-aromatic conditions. The p=0.038 is marginally significant and should be interpreted cautiously at this sample size.
- No multiple testing correction on the 13 per-condition comparisons (NB04, cell 14). Given the small number of tests, this is tolerable but should be acknowledged.
Co-fitness confidence thresholds (NB03, cell 11): The scheme (High: r>0.7 and gap>0.15; Medium: r>0.5; Low: r>0.3) produces 2 High and 14 Medium assignments. One consequence: ACIAD3137 (r=0.988 with Complex I, the highest correlation of any unknown gene) gets only "Medium" confidence because the gap to Aromatic pathway (r=0.890) is only 0.098, falling below the 0.15 gap threshold. This is arguably too conservative for genes with r>0.98. With only 8 growth conditions, many genes will correlate with multiple subsystems simply because they share the quinate defect -- the gap criterion addresses this but its specific thresholds aren't empirically justified.
Operon prediction (NB02, cell 6): The heuristic (<100 bp intergenic distance, same strand, allowing overlaps to -50 bp) is standard and appropriate. The Complex I operon prediction (13 genes, nuoA-N) and pca/qui operon (12 genes) match published data (Dal et al. 2005).
Pitfall awareness: The project correctly handles string-typed FB columns. Since most work is done locally on SQLite, most BERDL-specific pitfalls don't apply. Two project-specific pitfalls were captured in docs/pitfalls.md: keyword categorization fragility (line 733) and NotebookEdit cell validation issues (line 739).
Findings Assessment
Conclusions well-supported by data:
- The 4-subsystem model is convincingly supported by convergent evidence: FBA flux ratios, genomic independence (distinct chromosomal loci), and very high within-subsystem co-fitness (Complex I: r=0.992, Aromatic pathway: r=0.961).
- The FBA blind spot finding (30/51 genes with no reaction mappings, 0% predicted essentiality for Complex I despite 1.76x higher flux on aromatics) is clearly demonstrated and is a genuinely useful finding for metabolic modeling.
- Genomic independence of subsystems (NB02) is cleanly shown: the only cross-category operon involves Aromatic pathway + "Other" (ACIAD1710, which is actually a misclassified pathway gene).
- The cross-species finding that Complex I defects are largest on acetate (-1.55 deficit) and succinate (-1.39), not on aromatic substrates, is an important refinement. The NDH-2 compensation hypothesis is plausible and well-reasoned.
Areas of concern:
- Modest overall within/between co-fitness separation: NB03 cell 5 shows within-category mean r=0.552 vs between-category r=0.518. This is driven by the high correlations within Complex I and Aromatic pathway (operon co-regulation), while Other (r=0.425) and Unknown (r=0.342) categories have low internal correlations. The subsystem assignments for unknown genes should be interpreted with this context.
- Thin PQQ/Iron cross-species data: NB04 shows only 4 aromatic fitness entries from 1 gene in the PQQ/Iron category. The REPORT wisely focuses on Complex I, but this gap limits the cross-species validation of the PQQ and iron dependency claims.
- Small n for interaction test: The key interaction test (NB04, cell 12) -- whether Complex I's aromatic deficit exceeds background -- uses only n=5 Complex I genes. The p=0.038 is marginal and would not survive conservative multiple testing correction.
- Count evolution is clear but could be highlighted: The numbers shift from NB01 (10 Complex I, 4 Iron, 6 Aromatic) to the REPORT (21 Complex I, 7 Iron, 8 Aromatic) after co-fitness reassignment. NB03 cell 13 shows the before/after table, but a reader going notebook-by-notebook needs to track this transition carefully.
Limitations honestly acknowledged: The REPORT.md Limitations section identifies four appropriate caveats: limited 8-condition dimensionality, mixed cross-species signals, phenotypic-only evidence for the 11 newly assigned Complex I genes, and PQQ's shared glucose dependency. The PQQ-glucose point is particularly valuable as it moderates the aromatic-exclusivity claim.
No incomplete or placeholder analysis: All planned analyses were either completed or explicitly deferred with rationale to Future Directions.
Suggestions
-
Import
categorize_gene()from the existingutils.pyinstead of duplicating it (high impact, easy fix): Thenotebooks/utils.pymodule already contains a corrected version with "muconolactone" in the keyword list. Replace the inline function definitions in NB01 (cell 1), NB02 (cell 1), and NB03 (cell 1) withfrom utils import categorize_gene. This fixes the ACIAD1710 misclassification in the initial categorization and eliminates 3 copies of identical code. -
Acknowledge the small sample size in the interaction test (high impact, easy fix): NB04's interaction test (cell 12) uses only n=5 Complex I genes and yields p=0.038. Add a sentence noting this marginal significance and small n. The per-condition analysis (cell 14) provides complementary evidence, but the formal interaction claim should be appropriately hedged. Consider a permutation-based test as a robustness check.
-
Document the ortholog transfer methodology in NB04 (medium impact): NB04 cell 3 briefly mentions BBH ortholog detection, but the analysis relies entirely on pre-computed
fitness_avgandfitness_countvalues from the SQLite database. Clarify: how many FB organisms contributed to eachfitness_avgvalue? Were scores averaged across all organisms or only the best hit? This is important because aggregating fitness across organisms with different respiratory chain architectures (a limitation the REPORT itself notes) makes the individual values hard to interpret. -
Add the "muconolactone" keyword to the notebook-local categorize functions (medium impact, immediate fix): If refactoring to import from
utils.pyis deferred, at minimum add "muconolactone" to the keyword list in the notebook copies so ACIAD1710 (pcaC) is correctly classified as "Aromatic pathway" from the start. The current misclassification means NB01 and NB02 analyses (FBA comparison, operon analysis) undercount the aromatic pathway by 1 gene. -
Provide rationale for co-fitness confidence thresholds (medium impact): The gap>0.15 requirement for "High" confidence (NB03, cell 11) causes genes with r>0.98 to be classified as only "Medium." Consider whether the gap threshold should scale with the absolute correlation level -- e.g., a gene with r=0.988 to its best match might reasonably be "High" confidence even with a gap of 0.098, since the correlation is so extreme.
-
Add context for the modest overall within/between co-fitness difference (low-medium impact): A brief note in NB03 explaining that the overall within vs between difference (0.552 vs 0.518) is small because it aggregates tight subsystems (Complex I, Aromatic pathway) with heterogeneous categories (Other, Unknown) would help readers correctly interpret the co-fitness assignments.
-
Consider Bonferroni or BH-FDR correction for per-condition comparisons (low impact, easy fix): NB04 cell 14 makes 13 condition-level comparisons. Even with a lenient BH-FDR threshold, this would strengthen the claim that acetate and succinate show the largest disproportionate Complex I defects.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Chromosome Map
Cofitness Heatmap
Cofitness Within Between
Complex I Vs Background
Cross Species Fitness
Fba Flux Heatmap
Gene Clusters
Support Network Categories
Unknown Assignments Heatmap