Fitness Cost of Antimicrobial Resistance Genes
CompletedResearch Question
Do antimicrobial resistance (AMR) genes impose a fitness cost in the absence of antibiotic selection pressure? Using genome-wide RB-TnSeq fitness data from 28 bacteria, we test whether transposon knockouts of AMR genes show systematically positive fitness (mutant grows better than wildtype) under standard growth conditions, indicating the intact AMR gene is a metabolic burden.
Overview
This project combines three BERDL data assets to test the "cost of resistance" hypothesis at pangenome scale:
- bakta_amr (83K AMR gene clusters) identifies resistance genes across 132.5M pangenome clusters
- Fitness Browser (48 organisms, 27M fitness measurements) provides genome-wide transposon fitness data
- fb_pangenome_link (177K gene-to-cluster links) bridges fitness browser genes to pangenome clusters
We identify 1,352 AMR genes across 28 organisms with fitness data, compute their fitness under non-antibiotic conditions, and compare to non-AMR background using a random-effects meta-analysis. Validation uses antibiotic experiments as positive controls.
Key Findings
1. Universal cost of resistance across 25 bacterial species (H1 supported)
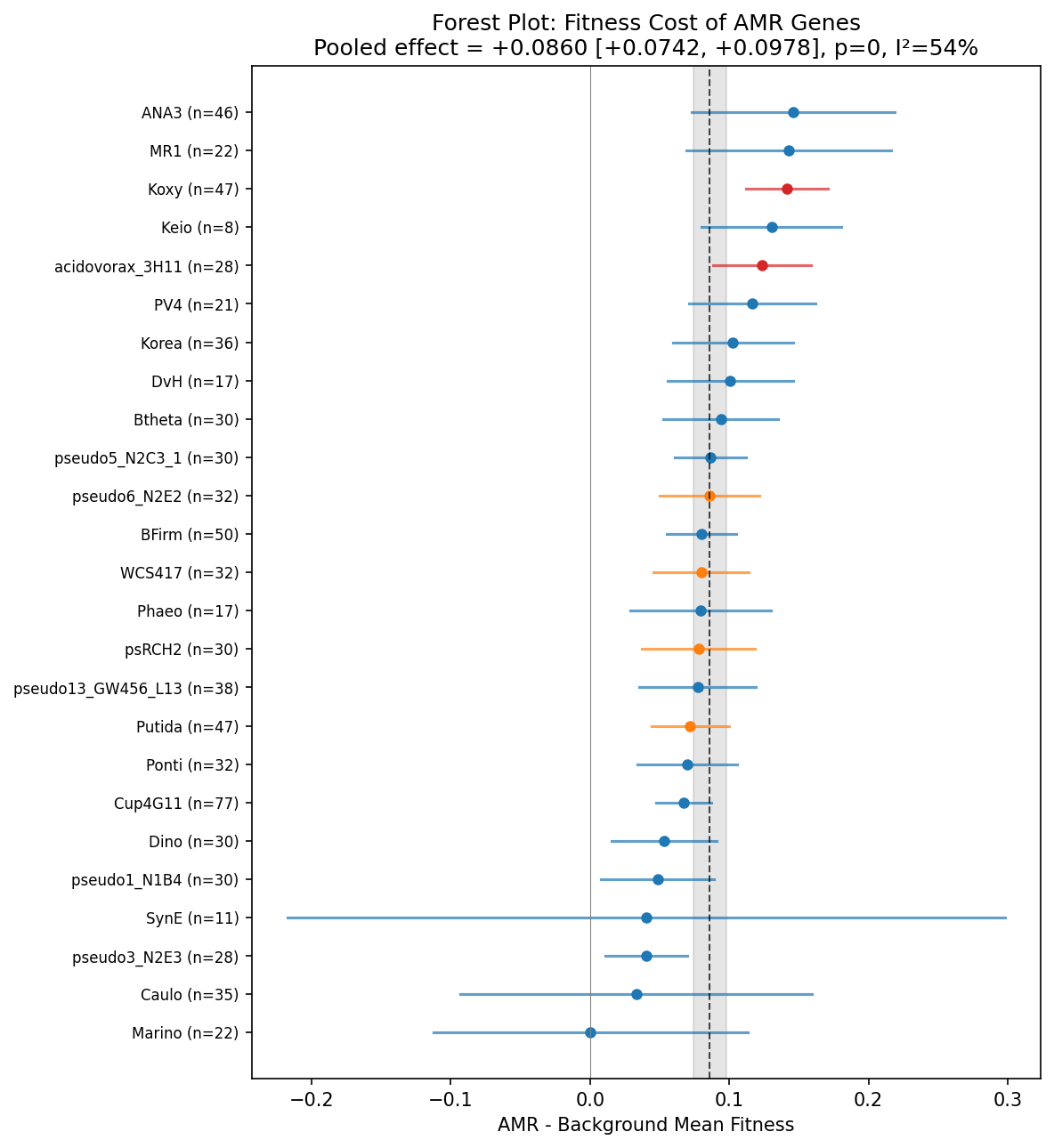
AMR gene knockouts show systematically higher fitness than non-AMR gene knockouts under non-antibiotic conditions, confirming that resistance genes impose a metabolic burden. A DerSimonian-Laird random-effects meta-analysis across 25 organisms yields a pooled effect of +0.086 [95% CI: +0.074, +0.098], z = 14.3, p ~ 0. Remarkably, all 25 of 25 organisms show a positive shift (AMR > background), making this one of the most consistent signals observed across the Fitness Browser compendium. The median per-organism Cohen's d = 0.18, indicating a small but real effect — consistent with the literature prediction of +0.05 to +0.20 for fitness costs after compensatory evolution.
Only 4.6% of AMR genes were absent from the fitness matrices (putatively essential), substantially lower than the ~14% background essential rate estimated in a prior analysis (fitness_effects_conservation, different organism set). AMR genes being less likely to be essential is consistent with the cost-of-resistance framework — these genes are more dispensable, not less. The low essential rate also argues against strong right-censoring bias.
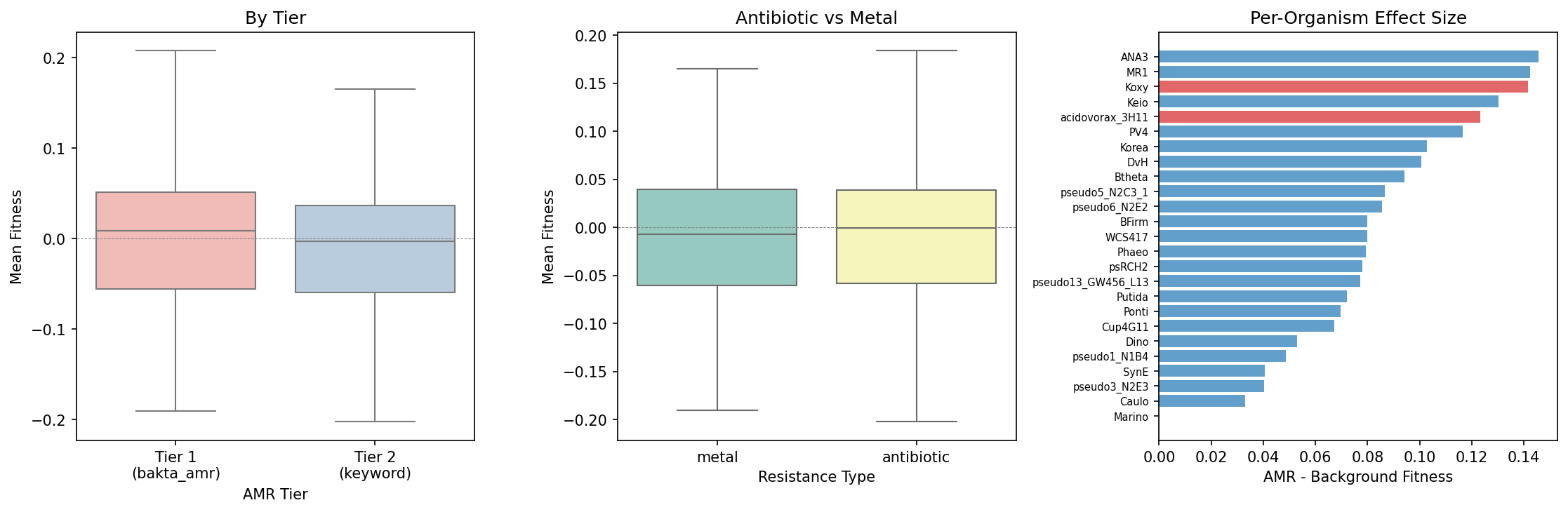
The Tier 1 (bakta_amr, N=110) and Tier 2 (keyword annotation, N=691) gene sets show indistinguishable fitness distributions (KS p = 0.17), confirming that the keyword-based Tier 2 expansion does not dilute or bias the signal.
(Notebook: 02_fitness_cost_analysis.ipynb)
2. AMR genes become more important under antibiotic pressure (H4 partially supported)
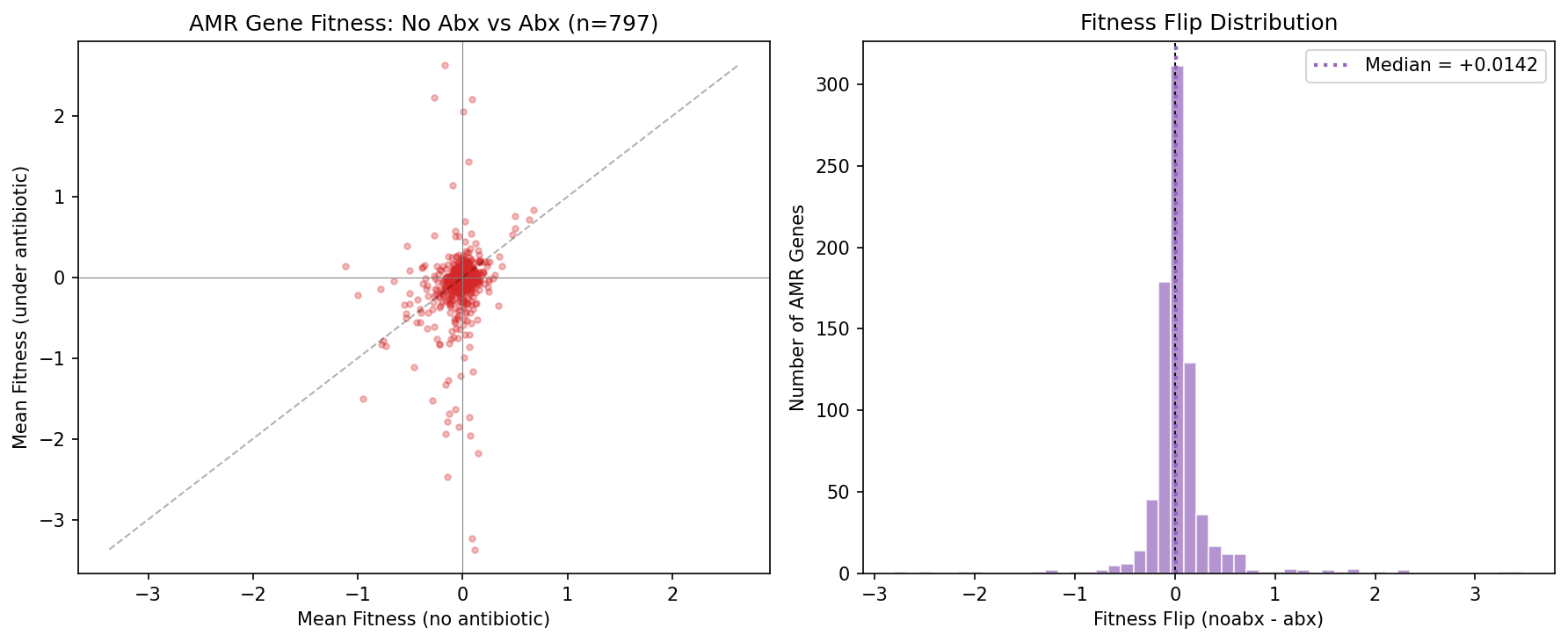
When tested under any antibiotic, 57% of AMR genes show a fitness flip — they become relatively more important (lower fitness when knocked out) compared to non-antibiotic conditions (Wilcoxon signed-rank p = 0.0001, N = 797). The mean flip is +0.045 (noabx − abx fitness), indicating AMR genes shift from slight burden to slight importance.
Critically, this flip is mechanism-dependent: efflux genes (broad-spectrum) show a significantly stronger flip than enzymatic inactivation genes (narrow-spectrum): +0.094 vs −0.001, MWU p = 0.007. This is expected — broad-spectrum efflux pumps protect against many antibiotics and should show fitness importance under any antibiotic experiment, while narrow-spectrum enzymes (e.g., beta-lactamases) only become important when the matching antibiotic is present. The 43% of AMR genes that don't flip likely reflect narrow-spectrum resistance being tested against non-matching antibiotics, not non-functional genes.
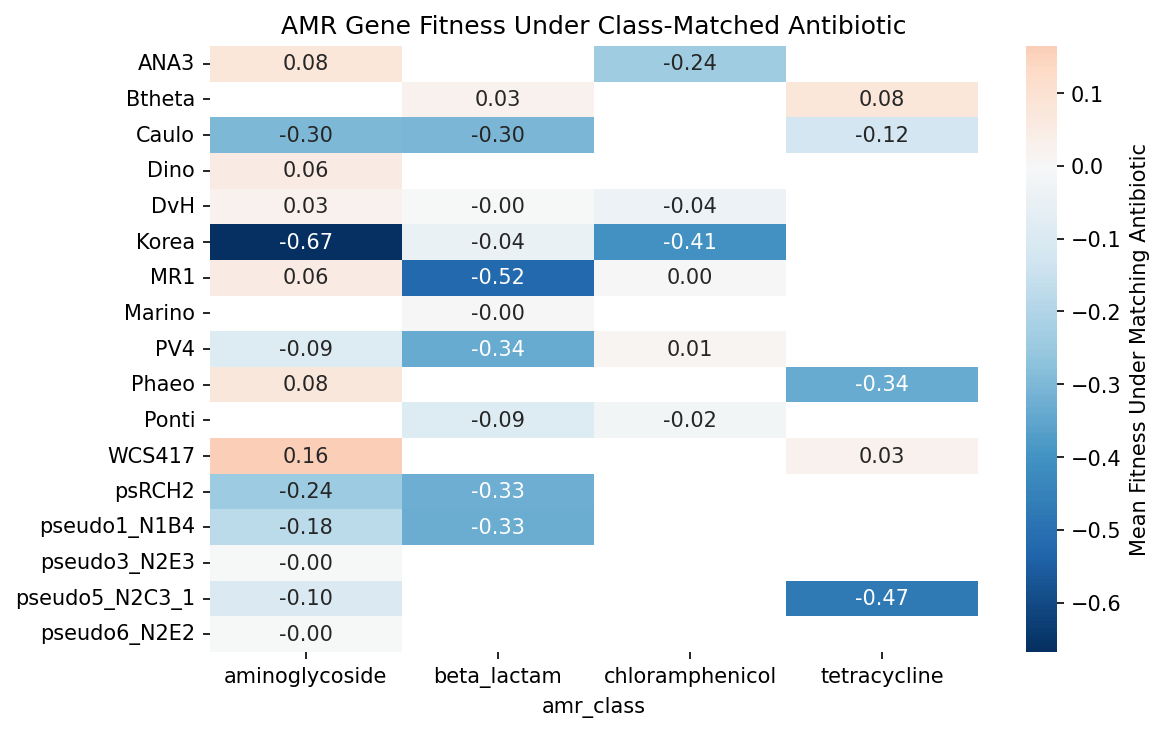
The class-matched validation (157 gene-antibiotic pairs across 4 resistance classes) shows a mean flip of +0.113, but the Wilcoxon signed-rank test does not reach significance (p = 0.14). This is likely due to the small N per class after matching and heterogeneity across organisms — some organisms show strong flips while others are near zero. The any-antibiotic analysis (N = 797) has much greater power and is highly significant. Within the class-matched set, chloramphenicol resistance genes show the strongest validation: 6/6 (100%) show the expected flip. Beta-lactam genes (105 pairs across 10 organisms) show 50% flip rate, consistent with many being tested against non-carbenicillin beta-lactams.
(Notebooks: 03_antibiotic_validation.ipynb, 04b_followup_analyses.ipynb)
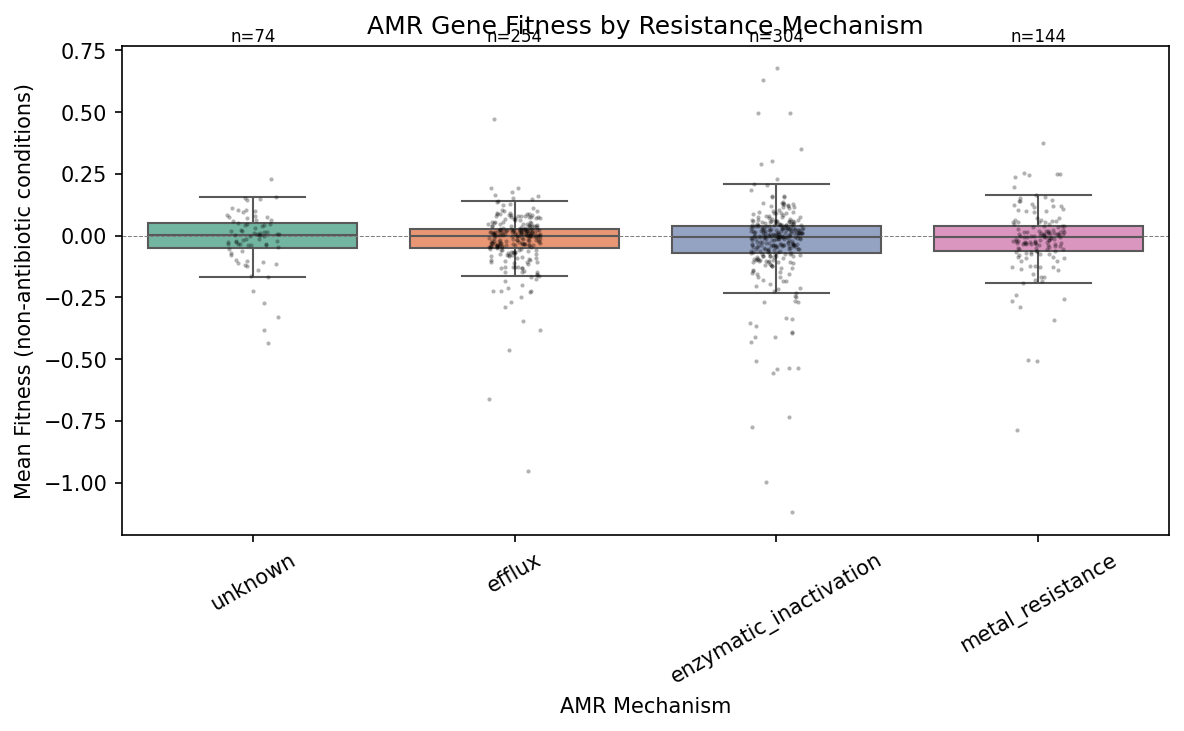
3. Resistance mechanism does not predict fitness cost (H2 not supported)
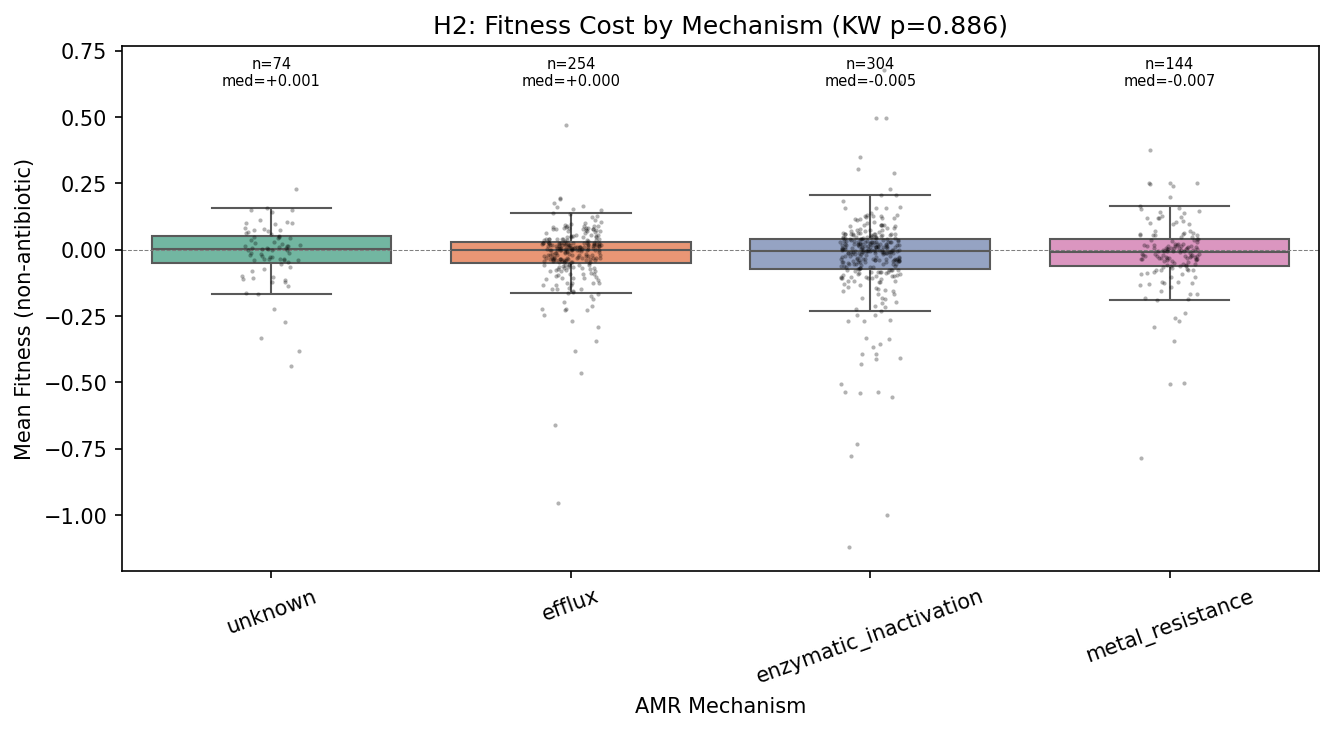
Contrary to H2, the fitness cost of AMR genes does not vary by resistance mechanism. The Kruskal-Wallis test across four testable mechanisms (efflux N=254, enzymatic inactivation N=304, metal resistance N=144, unknown N=74) is not significant (H = 0.65, p = 0.89). The Jonckheere-Terpstra test for the predicted ordering (efflux > enzymatic > metal > unknown) is also non-significant (z = 0.23, p = 0.41). No pairwise comparison survives FDR correction.
This uniformity is itself a finding. If all resistance mechanisms impose the same modest cost (~0.086 fitness units relative to background), it suggests that only AMR genes whose cost has been minimized through compensatory evolution persist in these genomes. The RB-TnSeq organisms are lab-adapted strains that have had extensive evolutionary time to compensate for resistance costs. The uniform cost may represent a "floor" — the irreducible metabolic overhead of maintaining any extra gene, regardless of its function.
(Notebook: 04_stratification.ipynb)
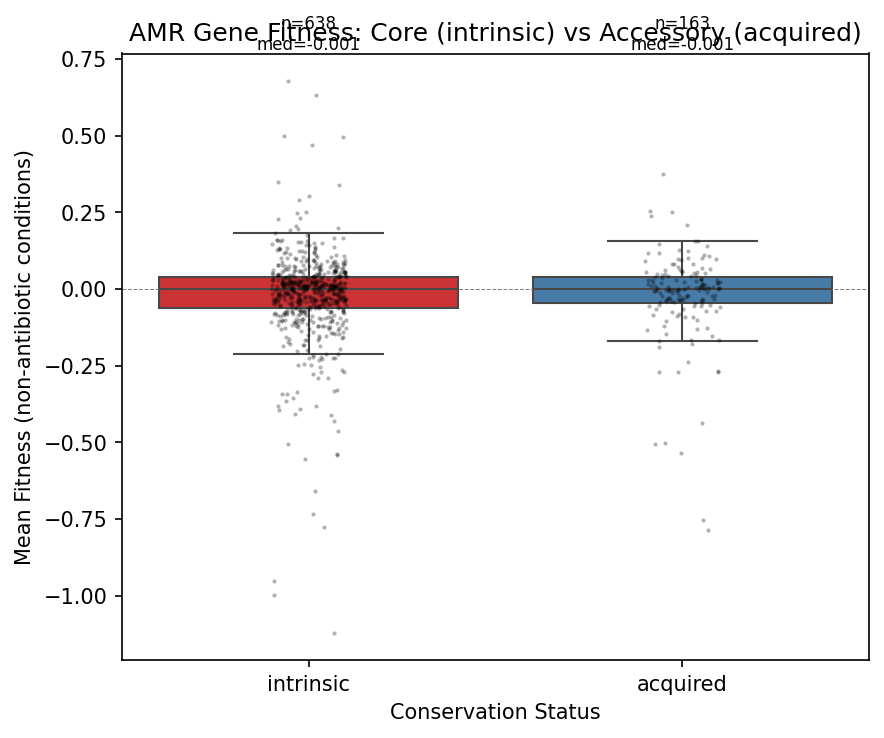
4. Core and accessory AMR genes have identical fitness costs (H3 not supported)
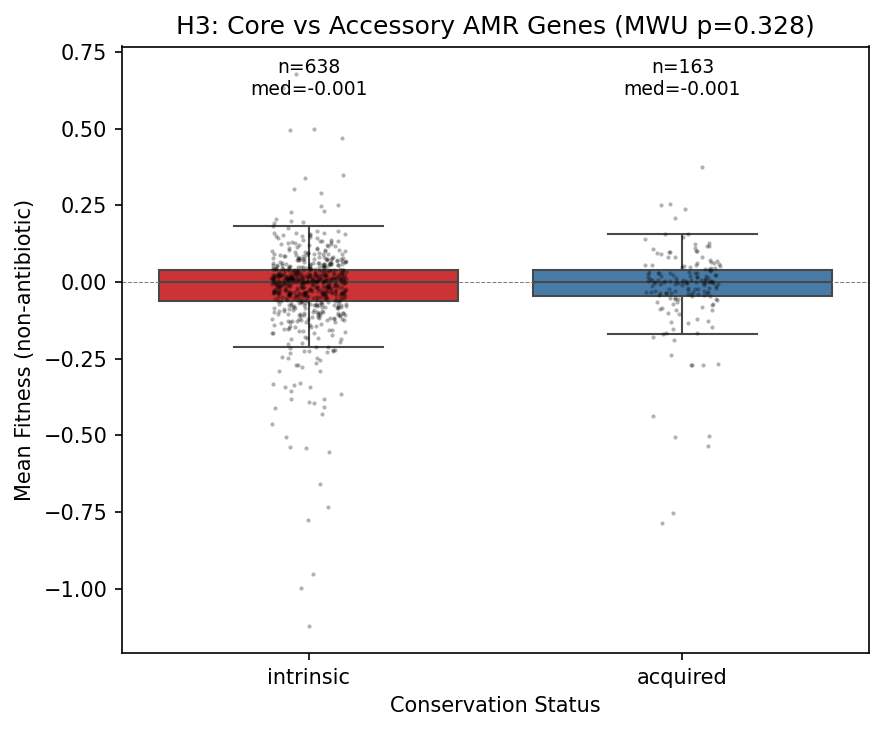
Core (intrinsic, N=638) and accessory (acquired, N=163) AMR genes show virtually identical fitness distributions: mean −0.024 vs −0.024, Cohen's d = 0.002, MWU p = 0.33.
An important caveat: most FB species have few genomes in GTDB (median 9, range 2–399), which makes the ≥95% prevalence threshold for "core" designation imprecise. With only 2–9 genomes, a gene present in all 9 is labeled "core" but could be accessory at larger sampling depth. However, this imprecision should add noise (diluting any real difference), not create a false null — so the absence of a difference is likely genuine, at least for the well-sampled species (e.g., K. michiganensis with 399, B. thetaiotaomicron with 287, S. meliloti with 241 genomes).
The result challenges the expectation that recently acquired (accessory) AMR genes should be costlier because they haven't been optimized. One interpretation is that acquisition of AMR genes is not random — horizontal transfer preferentially captures genes that have already been cost-optimized in their donor lineage, or that the receiving genome rapidly compensates.
(Notebooks: 04_stratification.ipynb, 04b_followup_analyses.ipynb)
5. Mechanism is strongly associated with conservation, even though cost is not
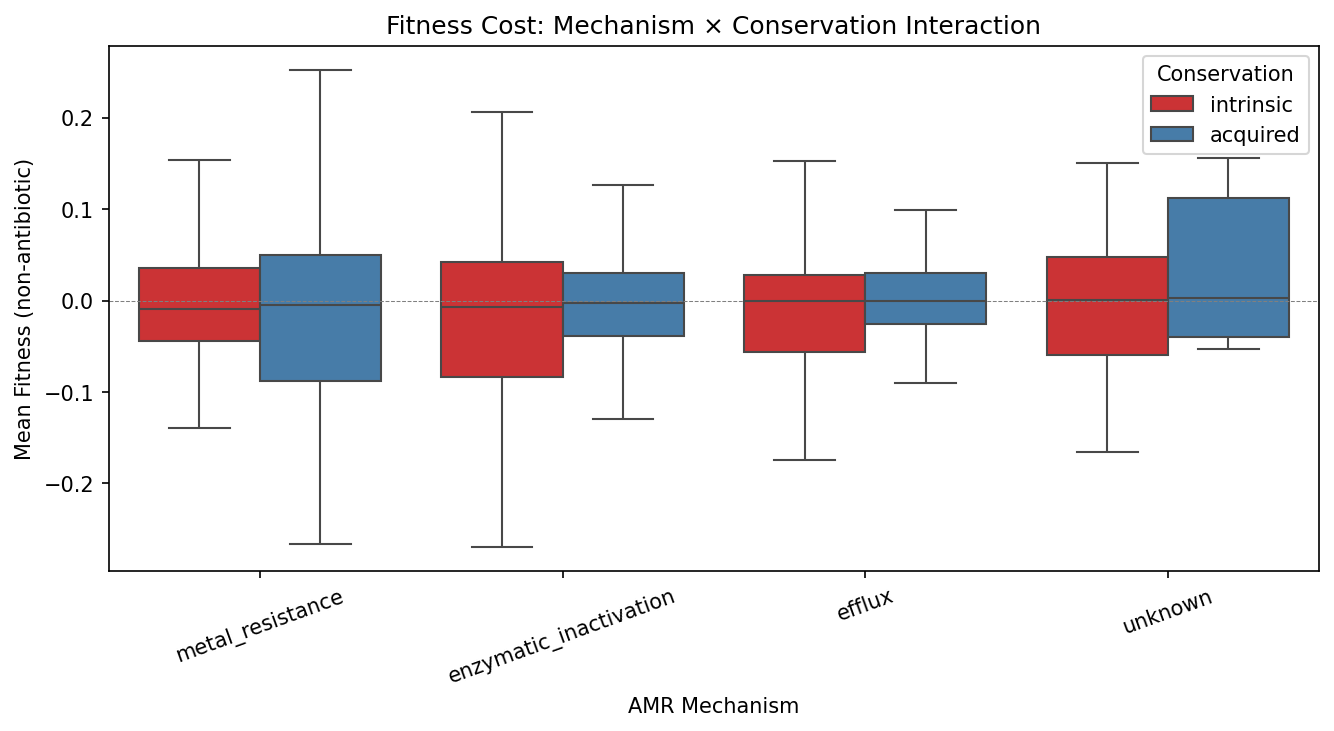
While mechanism doesn't predict fitness cost, it strongly predicts conservation status (χ² = 69.3, p = 1.4×10⁻¹³). Metal resistance genes are 44% accessory, while efflux (13%) and enzymatic inactivation (16%) genes are overwhelmingly core. This decoupling — mechanism predicts where in the pangenome an AMR gene sits, but not how costly it is — suggests that the forces governing AMR gene retention (horizontal transfer frequency, selective advantage in specific environments) are distinct from the forces governing metabolic cost.
(Notebook: 04b_followup_analyses.ipynb)
Results
Data Assembly (NB01)
- 1,352 AMR genes identified across 43 organisms (178 Tier 1, 1,174 Tier 2)
- 28 organisms have both AMR genes and fitness matrices; 25 qualify for per-organism tests (≥5 AMR genes)
- 6,804 experiments classified: 2,868 carbon/nitrogen, 1,862 stress, 727 standard, 447 metal, 443 antibiotic
- AMR class distribution: beta-lactam (44 T1), mercury (27), arsenic (22), efflux_rnd (22)
Per-Organism Effect Sizes (NB02)
| Metric | Value |
|---|---|
| Organisms tested | 25 |
| Organisms with positive shift | 25/25 (100%) |
| Nominally significant (p<0.05) | 6 |
| FDR significant (q<0.05) | 2 (acidovorax_3H11, Koxy) |
| Pooled effect (DL random-effects) | +0.086 [+0.074, +0.098] |
| I² heterogeneity | 54.3% |
| Cochran's Q | 52.54, p = 0.0007 |
| Median Cohen's d | 0.18 |
Antibiotic Validation (NB03)
| Metric | Class-Matched | Any-Antibiotic |
|---|---|---|
| Gene-antibiotic pairs | 157 | 797 |
| Flip rate (more important under abx) | 54.8% | 57.0% |
| Mean fitness flip | +0.113 | +0.045 |
| Wilcoxon p (flip > 0) | 0.14 | 0.0001 |
Stratification (NB04)
| Stratification | Groups | Test | p-value | Result |
|---|---|---|---|---|
| Mechanism | 4 groups (N≥20) | Kruskal-Wallis | 0.89 | No difference |
| Mechanism (ordered) | efflux > enzymatic > metal | Jonckheere-Terpstra | 0.41 | No trend |
| Conservation | core vs accessory | Mann-Whitney U | 0.33 | No difference |
| Tier | T1 vs T2 | Mann-Whitney U | 0.26 | No difference |
| Resistance type | antibiotic vs metal | Mann-Whitney U | 0.87 | No difference |
| Mechanism × conservation | 6 groups | Chi-square | 1.4×10⁻¹³ | Strongly associated |
Interpretation
The cost of resistance is universal but uniform
The central finding is that AMR genes impose a small, consistent metabolic burden across all 25 tested bacterial species, all resistance mechanisms, and both core and accessory gene pools. The pooled effect of +0.086 means that AMR gene knockouts are 0.086 fitness units less detrimental than the average gene knockout — i.e., AMR genes are more dispensable than the typical gene. Note that absolute AMR knockout fitness averages −0.024 (slightly below the pool mean), so AMR knockouts still grow slower than the pool average; the cost manifests as a relative difference from the non-AMR background (which averages around −0.11). In practical terms, a cell that loses an AMR gene gains a small competitive advantage over cells that lose a randomly chosen gene, enough to matter over evolutionary timescales but invisible in short-term growth assays. This reconciles two apparently contradictory observations in the AMR literature: fitness costs are real (supporting resistance decline under stewardship), but they are small (explaining why resistance persists long after antibiotic withdrawal).
A note on the one-sample test: A Wilcoxon signed-rank test asking whether AMR knockout fitness is > 0 (i.e., whether AMR knockouts grow faster than the pool wildtype) gives p = 0.999, strongly rejecting that framing. This is expected — AMR gene knockouts, like most gene knockouts, grow slightly slower than wildtype. The relevant question is not "are AMR knockouts beneficial?" but "are AMR knockouts less costly than typical knockouts?" — which the per-organism Mann-Whitney design directly tests. The +0.086 effect captures this relative advantage.
Broad-spectrum resistance shows antibiotic-dependent importance
The validation analysis reveals a previously undescribed pattern: the fitness flip under antibiotic exposure is mechanism-dependent even though the baseline cost is not. Efflux pumps (broad-spectrum) become significantly more important under any antibiotic (flip +0.094), while enzymatic inactivation genes (narrow-spectrum) only flip under their matching antibiotic. This decoupling of cost-independence and benefit-specificity has implications for predicting which resistance mechanisms will be maintained in antibiotic-free environments.
Conservation reflects acquisition history, not fitness cost
The strong association between mechanism and conservation (metal resistance: 44% accessory vs efflux: 13%) combined with the absence of a cost difference between core and accessory genes suggests that AMR gene conservation reflects how and when genes were acquired rather than how much they cost. Efflux systems, often chromosomally encoded as part of general stress response, are maintained as core genes. Metal resistance operons, frequently carried on mobile elements, remain accessory — but both impose the same modest cost once established in a genome.
Literature Context
- Melnyk et al. (2015) meta-analyzed ~600 resistance cost measurements and found costs in ~70% of cases with a mean relative fitness of 0.91–0.95 (5–9% cost). Their measurements are absolute fitness differences between isogenic resistant and sensitive strain pairs, while our +0.086 is a relative difference between AMR and non-AMR transposon knockouts within pooled libraries — these are different measurement scales. Nonetheless, both converge on the same order of magnitude (small single-digit percent costs), providing reassuring concordance between the two methodologies.
- Andersson & Hughes (2010) predicted that compensatory mutations would reduce or eliminate fitness costs over time, making resistance reversal unlikely. Our finding that core and accessory AMR genes show identical costs is consistent with rapid compensatory evolution, even for recently acquired genes.
- Vanacker, Lenuzza & Rasigade (2023) meta-analyzed fitness costs in E. coli and found that horizontally transferred resistance genes (plasmid-borne) are less costly than chromosomal mutations. This explains why our transposon-based cost estimate is modest — many FB AMR genes are likely acquired determinants (beta-lactamases, acetyltransferases) rather than costly target modifications.
- Vogwill & MacLean (2015) found little systematic variation by mechanism after controlling for confounders, and emphasized environment-dependence. We confirm both: no mechanism-dependence in baseline cost, but clear environment-dependence (antibiotic validation).
- Olivares Pacheco & Alvarez (2017) showed that efflux pump costs in P. aeruginosa are metabolic (proton motive force drain) and rapidly compensated through metabolic rewiring. This explains why our efflux genes show the same modest cost as other mechanisms — compensation is a "general outcome."
- Levin, Perrot & Walker (2000) demonstrated that compensatory mutations arise faster than reversion to susceptibility, predicting that organisms maintain resistance at reduced cost. Our +0.086 may represent the irreducible cost after compensatory evolution — the minimum overhead of expressing a functional protein that provides no benefit without antibiotics.
- Roux et al. (2015) showed that resistance genes (particularly efflux pumps) can be beneficial in vivo by exporting host antimicrobials. This caveat applies: our in vitro cost measurement may overestimate the true ecological cost.
- Durão, Balbontín & Gordo (2018) reviewed epistasis as a driver of cost variation across genetic backgrounds. Our consistent signal across 25 diverse organisms suggests the cost is robust across genetic backgrounds, which is notable given the role of epistasis.
Novel Contribution
This is the first pan-bacterial meta-analysis of AMR fitness costs using genome-wide transposon fitness data. Prior studies typically compare single resistant vs sensitive strain pairs in one organism. By leveraging 27M fitness measurements across 25 diverse bacteria, we can:
1. Demonstrate the universality of the cost (25/25 organisms positive)
2. Show that cost is mechanism-independent (Kruskal-Wallis p = 0.89)
3. Discover that broad-spectrum mechanisms show stronger antibiotic-dependent importance than narrow-spectrum ones (MWU p = 0.007)
4. Demonstrate that mechanism predicts genomic location (core vs accessory) but not fitness cost — a decoupling not previously observed
Limitations
- Lab adaptation bias: All 25 organisms are lab-adapted strains. Compensatory evolution during laboratory maintenance may have reduced measurable costs below what wild strains experience. This makes our positive result more convincing but may underestimate the true cost in natural populations.
- Tier 2 annotation noise: 86% of AMR genes are Tier 2 (keyword-matched from bakta annotations), which may include some non-AMR genes (e.g., general efflux transporters). The Tier 1 sensitivity analysis shows consistent results, mitigating this concern.
- Antibiotic experiment coverage: Only 4 AMR classes (beta-lactam, aminoglycoside, chloramphenicol, tetracycline) have matched antibiotic experiments. Other classes (macrolide, glycopeptide, polymyxin) could not be validated.
- Essential gene censoring: ~4.6% of AMR genes are putatively essential (absent from fitness matrices). If these are the most costly AMR genes, our estimate of +0.086 is a lower bound.
- Relative, not absolute fitness: RB-TnSeq measures fitness relative to the pool average. The +0.086 is the difference between AMR knockout fitness (−0.024) and non-AMR knockout fitness (~−0.11), not an absolute selection coefficient. Comparisons to literature fitness costs measured by isogenic strain competition (e.g., Melnyk et al. 2015) are reassuring but not direct equivalences.
- Transposon insertion effects: Insertions can cause polar effects on downstream genes, potentially confounding fitness measurements for AMR genes in operons.
- Classification gaps: The Tier 2 product classifier does not handle fosfomycin resistance or tellurite resistance annotations, placing ~25 genes in the "unknown" mechanism category. Adding these would reclassify them to enzymatic_inactivation and metal_resistance respectively, slightly reducing the unknown group.
- Core/accessory label precision: Most FB species have few genomes (median 9), making the ≥95% prevalence threshold for "core" imprecise. The core vs accessory null result should be interpreted cautiously for species with <20 genomes.
Future Directions
- Within-organism mechanism subanalysis: For organisms with many AMR genes (Cup4G11: 77, BFirm: 50), test whether mechanism predicts cost within a single genetic background, controlling for phylogenetic differences.
- Efflux pump subclassification: Distinguish narrow-spectrum drug pumps from general RND systems (AcrAB-TolC) and test whether constitutively expressed efflux systems have lower costs.
- Condition-specific costs: Instead of averaging across all non-antibiotic experiments, test whether AMR costs vary under different stress conditions (metal, osmotic, carbon limitation).
- Cross-reference with metal fitness atlas: The 144 metal resistance genes with fitness data could be cross-referenced against the
metal_fitness_atlasproject to test whether genes costly under standard conditions are protective under metal stress. - Extend to full pangenome: Scale the analysis from 25 FB organisms to all 293K BERDL genomes by predicting AMR cost from gene cluster conservation patterns.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
bakta_amr, bakta_annotations |
AMR gene identification (Tier 1 + Tier 2) |
kescience_fitnessbrowser |
genefitness, gene, experiment |
Fitness measurements, gene metadata, experiment classification |
| Cross-project | conservation_vs_fitness/fb_pangenome_link.tsv |
Bridge FB genes to pangenome clusters |
| Cross-project | fitness_modules/matrices/* |
Pre-cached fitness matrices |
Generated Data
| File | Rows | Description |
|---|---|---|
data/amr_genes_fb.csv |
1,352 | AMR genes with class, mechanism, conservation, tier |
data/experiment_classification.csv |
6,804 | Experiments classified by type |
data/amr_fitness_noabx.csv |
801 | Per-gene mean fitness under non-antibiotic conditions |
data/organism_effect_sizes.csv |
25 | Per-organism effect sizes with CIs and p-values |
data/amr_fitness_abx_validation.csv |
954 | Fitness under antibiotic conditions (class-matched + any-antibiotic) |
data/amr_fitness_stratified.csv |
10 | Stratification summary by mechanism, conservation, tier, resistance type |
References
- Andersson DI, Hughes D. (2010). "Antibiotic resistance and its cost: is it possible to reverse resistance?" Nature Reviews Microbiology 8(4):260-71. PMID: 20208551
- Arkin AP, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nature Biotechnology 36(7):566-569. PMID: 29979655
- Bjorkman J, Nagaev I, Berg OG, Hughes D, Andersson DI. (2000). "Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance." Science 287(5457):1479-82. PMID: 10688795
- Durão P, Balbontín R, Gordo I. (2018). "Evolutionary mechanisms shaping the maintenance of antibiotic resistance." Trends in Microbiology 26(8):677-691. PMID: 29439838
- Levin BR, Perrot V, Walker N. (2000). "Compensatory mutations, antibiotic resistance and the population genetics of adaptive evolution in bacteria." Genetics 154(3):985-997. PMID: 10757748
- Melnyk AH, Wong A, Kassen R. (2015). "The fitness costs of antibiotic resistance mutations." Evolutionary Applications 8(3):273-83. PMID: 25861385
- Olivares Pacheco J, Alvarez-Ortega C, Alcalde Rico M, Martínez JL. (2017). "Metabolic compensation of fitness costs is a general outcome for antibiotic-resistant Pseudomonas aeruginosa mutants overexpressing efflux pumps." mBio 8(4):e00500-17. PMID: 28765215
- Price MN, et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557(7706):503-509. PMID: 29769716
- Roux D, Danilchanka O, Guillard T, et al. (2015). "Fitness cost of antibiotic susceptibility during bacterial infection." Science Translational Medicine 7(297):297ra114. PMID: 26203082
- San Millan A, MacLean RC. (2017). "Fitness costs of plasmids: a limit to plasmid transmission." Microbiology Spectrum 5(5). PMID: 28944751
- Vanacker M, Lenuzza N, Rasigade JP. (2023). "The fitness cost of horizontally transferred and mutational antimicrobial resistance in Escherichia coli." Frontiers in Microbiology 14:1186920. PMID: 37234529
- Vogwill T, MacLean RC. (2015). "The genetic basis of the fitness costs of antimicrobial resistance: a meta-analysis approach." Evolutionary Applications 8(3):284-95. PMID: 25861386
Discoveries
Random-effects meta-analysis of 801 AMR genes across 25 organisms shows a pooled fitness shift of +0.086 [+0.074, +0.098] when AMR genes are knocked out — all 25/25 organisms positive. This cost is mechanism-independent (KW p=0.89), conservation-independent (core = accessory, p=0.33), and tier-indep
Read more →Data Collections
Review
Summary
This is a well-designed, largely well-executed project that leverages 27M RB-TnSeq fitness measurements across 25 bacteria to test the cost-of-resistance hypothesis at pan-bacterial scale. The three-file project structure (README, RESEARCH_PLAN, REPORT) is used correctly; the RESEARCH_PLAN demonstrates genuine iterative refinement across three documented revisions including a NB02 checkpoint; statistical methods — DerSimonian-Laird random-effects meta-analysis, Jonckheere-Terpstra ordered-alternative test, pre-registered minimum group sizes — are appropriate and correctly implemented; and the central finding (25/25 organisms show a positive AMR-vs-background fitness shift) is scientifically compelling and well-supported. Known pitfalls (locusId type mismatch, string-typed fitness values, essential gene censoring) are explicitly anticipated and correctly handled. Two issues require correction before this project is cited or built upon: requirements.txt is missing statsmodels, which is used in three notebooks and will cause an ImportError for anyone following the documented reproduction steps; and NB04b's genome-count sensitivity analysis silently fails because it uses linked gene count rather than genome count as a proxy — every species exceeds every threshold, making the three comparisons trivially identical and uninformative.
Methodology
Research question and hypotheses: The research question is clearly stated and operationally testable. All four hypotheses (H1–H4) and the null H0 have explicit, measurable predictions. Minimum group sizes are pre-registered (N≥5 for per-organism tests, N≥20 for mechanism tests). The checkpoint design between NB02 and NB03-04 is documented in the RESEARCH_PLAN, and the v3 revision records that the checkpoint was actually evaluated and the plan approved to proceed.
Unit of analysis: The per-gene mean fitness approach (averaging across experiments before running per-organism Mann-Whitney tests) avoids inflating the per-organism p-values by the number of experiments. This is methodologically correct and explicitly justified in both the RESEARCH_PLAN and REPORT.
Two-tier AMR identification: The Tier 1 (strict bakta_amr) / Tier 2 (keyword bakta_annotations) strategy is well-reasoned. The NB02 spot-check of 114 unclassified Tier 2 genes confirms they are genuinely AMR-related (fosfomycin resistance proteins, RND efflux MFP subunits, tellurite resistance proteins — no obvious false positives). The Tier 1–only sensitivity analysis (KS D=0.112, p=0.17) supports pooling the tiers.
Cross-project dependencies: Three prior project artifacts are consumed (conservation_vs_fitness/fb_pangenome_link.tsv, fitness_modules/matrices/, essential_genome/). These are clearly documented and their paths are resolved robustly. The ~14% background essential rate cited in the REPORT is correctly attributed to a different project (fitness_effects_conservation) using a different organism set; the REPORT notes the difference ("different organism set") and interprets the lower AMR essential rate (4.6%) correctly as consistent with AMR genes being more dispensable than average.
Reproducibility: The README reproduction section clearly lists execution order, identifies which step requires Spark (only src/extract_amr_data.py), and notes that all downstream notebooks can run locally from cached data. This is the correct pattern. However, requirements.txt lists only pandas, numpy, scipy, matplotlib, seaborn — it omits statsmodels, which is imported as from statsmodels.stats.multitest import multipletests in NB02 (cell-13), NB03 (cell-1), and NB04 (cell-5). Anyone following the documented reproduction steps will hit an ImportError at the first BH-FDR correction.
Code Quality
SQL and Spark (extract_amr_data.py): The batched IN-clause approach for querying 163K cluster IDs in blocks of 5,000 is correct and necessary. The import from berdl_notebook_utils.setup_spark_session import get_spark_session matches the JupyterHub CLI pattern documented in pitfalls.md. Keyword terms for Tier 2 extraction are specific enough to avoid metabolic enzymes (efflux pump hits are gated on rnd, multidrug, antibiotic, or resistance).
Known pitfall handling: Three pitfalls are explicitly anticipated in the RESEARCH_PLAN and correctly handled in the code:
- locusId integer vs. string mismatch: fit_mat.index = fit_mat.index.astype(str) and explicit str(l) casts throughout NB02–NB04 (NB02 cell-8).
- String-typed fitness values: fit_mat = fit_mat.apply(pd.to_numeric, errors='coerce') (NB02 cell-8).
- Essential gene censoring: quantified per organism in NB02 cell-6 (4.6% overall), with direction of bias discussed in the REPORT.
DerSimonian-Laird implementation (NB02 cell-15): Correct. Computes fixed-effects weights, Cochran's Q, τ², random-effects weights, pooled estimate, 95% CI using 1.96×SE, one-sided z-test for effect > 0. Output matches the REPORT (θ=+0.086, CI=[+0.074, +0.098], z=14.3, I²=54.3%).
Jonckheere-Terpstra implementation (NB04 cell-4): Conceptually correct but implemented as a nested double loop over all pairs across all four group combinations. With N=254 (efflux), N=304 (enzymatic inactivation), N=144 (metal), and N=74 (unknown), this iterates over roughly 254×304 + 254×144 + 254×74 + 304×144 + 304×74 + 144×74 ≈ 202K pair comparisons. Likely ran without timeout but the runtime could be several minutes on slow hardware. Consider noting the expected runtime or referencing a faster implementation.
AMR product classifier — fosfomycin and tellurite gap (NB01 cells 7 and 9): The SQL extraction in extract_amr_data.py correctly captures "Fosfomycin resistance protein AbaF" (3 genes, confirmed in NB01 cell-3 spot-check) and "Tellurite resistance protein" variants (>15 genes). However, classify_amr_product() in NB01 cell-7 has no matching branch for either fosfomycin or tellurite, so these ~25 genes propagate through all analyses as amr_class='unclassified', amr_mechanism='unknown'. They are counted in the unknown mechanism group (N=74 in NB04) alongside genuine unknowns. The REPORT's Limitations section acknowledges this gap, but it is fixable with two additional if branches.
NB04b cells 10–12 — genome-count sensitivity analysis (critical logic error): The code computes:
genome_counts = fb_link.groupby('gtdb_species_clade_id')['locusId'].count()
This counts linked genes per species (the fb_link table has one row per gene-cluster link). The resulting column n_genes_linked ranges from 1,020 to 7,187 — values that represent gene counts, not genome counts. Threshold comparisons (>=50 genomes, >=100 genomes, >=200 genomes) are then applied to this column. Since every species far exceeds even the highest threshold, all three comparisons include the full dataset identically: core N=638, accessory N=163, p=0.6562 at all three levels. The cell-12 output confirms this — mean=4,860, min=1,020. The intended sensitivity analysis (whether the H3 null result holds for well-sampled species with reliable core/accessory labels) was not executed. This is a silent failure — no error is raised, results look plausible, but they are uninformative.
Findings Assessment
H1 (universal cost of resistance): The 25/25 positive per-organism shift and meta-analytic pooled effect (+0.086 [+0.074, +0.098], z=14.3) are compelling. The REPORT correctly interprets this as a relative difference: "AMR gene knockouts are 0.086 fitness units less detrimental than the average gene knockout — i.e., AMR genes are more dispensable than the typical gene" and notes that "absolute AMR knockout fitness averages −0.024 (slightly below the pool mean), so AMR knockouts still grow slower than the pool average." The REPORT also correctly addresses the one-sample test (Wilcoxon p=0.9985 against zero), explaining that the relevant comparison is AMR vs. background knockouts within each organism, not AMR vs. wildtype. Both the effect size interpretation and the one-sample test discussion are accurate and transparent.
H4 (antibiotic validation): The any-antibiotic Wilcoxon (p=0.0001, N=797) provides solid positive-control support. The class-matched Wilcoxon (p=0.14, N=157) is non-significant. The REPORT correctly reports both results in the summary table and acknowledges the class-matched non-significance in the REPORT narrative with a plausible explanation (small N per class, heterogeneous experiments). The efflux vs. enzymatic flip comparison (NB04b cell-4, MWU p=0.007) is a genuinely interesting secondary finding that supports the narrow-spectrum vs. broad-spectrum interpretation.
H2 (mechanism-dependent costs): The null result is clearly reported (KW H=0.65, p=0.89; JT z=0.23, p=0.41) and all pairwise comparisons survive FDR correction at q>0.8. The interpretation — uniform compensatory evolution reaching a "floor" cost — is biologically plausible and supported by the cited literature (Olivares Pacheco et al. 2017; Vogwill & MacLean 2015).
H3 (accessory > core costs): The null result is clearly reported (MWU p=0.33, Cohen's d=0.002). The caveat about imprecise core/accessory labels for species with few genomes (median 9 per FB organism) is appropriately flagged. However, the genome-count sensitivity analysis intended to test this (NB04b cells 10–12) fails silently as described above, so the imprecision concern cannot actually be ruled out from the current analysis.
Mechanism × conservation interaction (NB04b cell-6): The chi-square result (χ²=69.3, p=1.4×10⁻¹³) is correctly computed and the finding — mechanism predicts pangenome location but not fitness cost — is genuinely novel and well-framed.
Literature comparisons: The comparison to Melnyk et al. (2015)'s 5–9% absolute fitness cost is honest about the methodological difference ("these are different measurement scales") while noting convergent order-of-magnitude agreement. The literature section is unusually thorough for a BERDL project (8 targeted citations, each linked to a specific finding).
Suggestions
-
(Critical) Add
statsmodelstorequirements.txt. It is imported in NB02 cell-13, NB03 cell-1, and NB04 cell-5 (from statsmodels.stats.multitest import multipletests) and will causeImportErrorfor any user who installs only the listed packages. -
(Critical) Fix or remove the genome-count sensitivity analysis in NB04b cells 10–12. The variable
n_genes_linkedholds gene counts (range 1,020–7,187), not genome counts, so all threshold comparisons are trivially identical. The correct fix is to retrieve actual genome counts from the pangenomepangenometable (no_genomescolumn) by joining ongtdb_species_clade_idvia a Spark query, or to query thespecies_genome_counts.csvfile that already exists in the project'sdata/directory (which may contain this information). If a Spark query is not feasible post-hoc, remove the cells and add a note to the REPORT Limitations that the genome-count precision analysis was not completed. -
(Moderate) Add fosfomycin and tellurite handling to
classify_amr_product()in NB01 cell-7. Two branches would reclassify ~25 currently-unknown genes:if 'fosfomycin' in product_lower→('fosfomycin', 'enzymatic_inactivation')andif 'tellurite' in product_lower→('tellurite', 'metal_resistance'). This would reduce the unexplainedunknownmechanism group from N=74 to approximately N=49 and eliminate the need for the Limitation 7 caveat in the REPORT. -
(Minor) Add a note in NB04b cell-3 clarifying why the "any_abx with class match" flip (mean=−0.006) differs from the class-matched flip (mean=+0.113). The difference arises because the "any_abx" set averages over all antibiotic experiments for each gene, diluting the signal from the single class-matching antibiotic. This is not an error, but the discrepancy is confusing and the MWU comparison (p=0.9772, class-matched flip vs. unmatched flip using any-abx fitness) tests an inconsistent pairing.
-
(Minor) Add a runtime comment to the Jonckheere-Terpstra implementation in NB04 cell-4. The O(n₁×n₂) pair-counting over four groups of 74–304 genes involves ~202K comparisons. It likely completed in under a minute, but noting the expected runtime would help future users who might apply it to larger datasets.
-
(Minor) Check
data/species_genome_counts.csv— this file exists in the projectdata/directory (visible in the file listing) but is never loaded or used in any notebook. If it contains per-species genome counts from the pangenome, it could directly fix Suggestion 2 without a Spark query.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
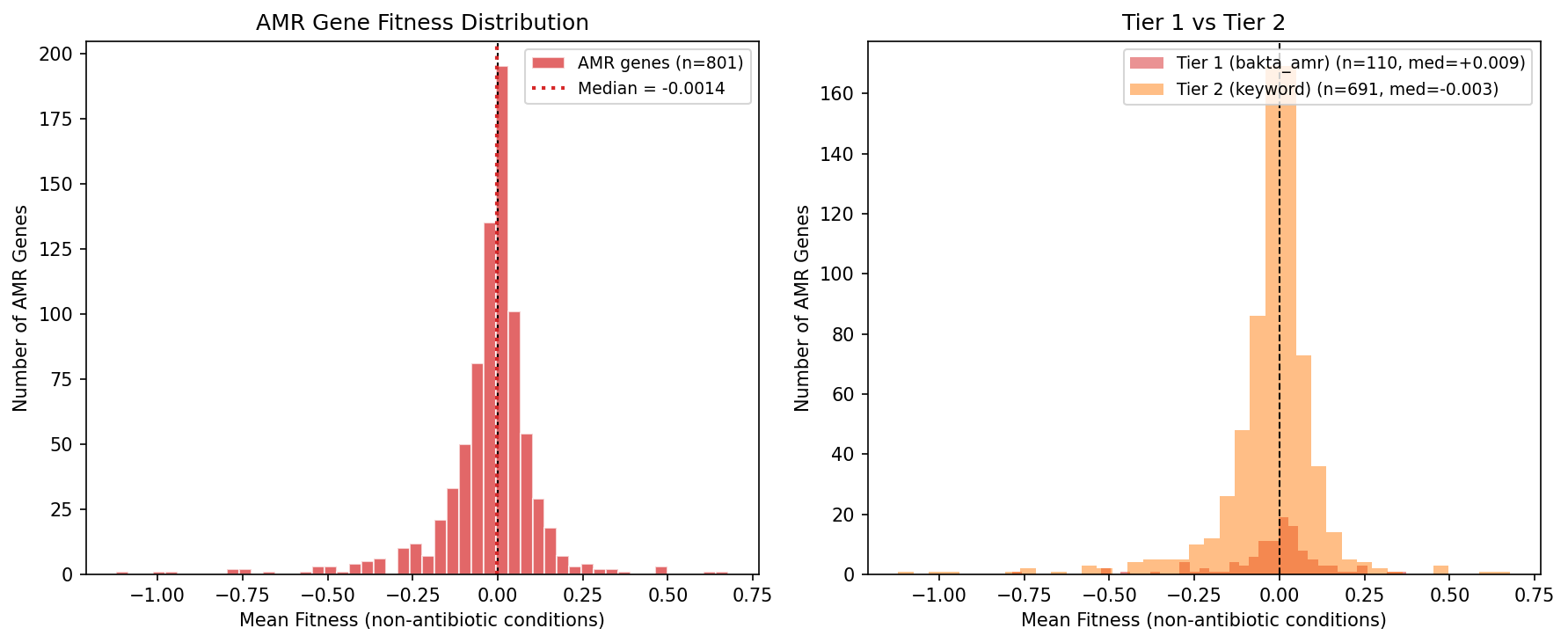
Amr Fitness Distribution
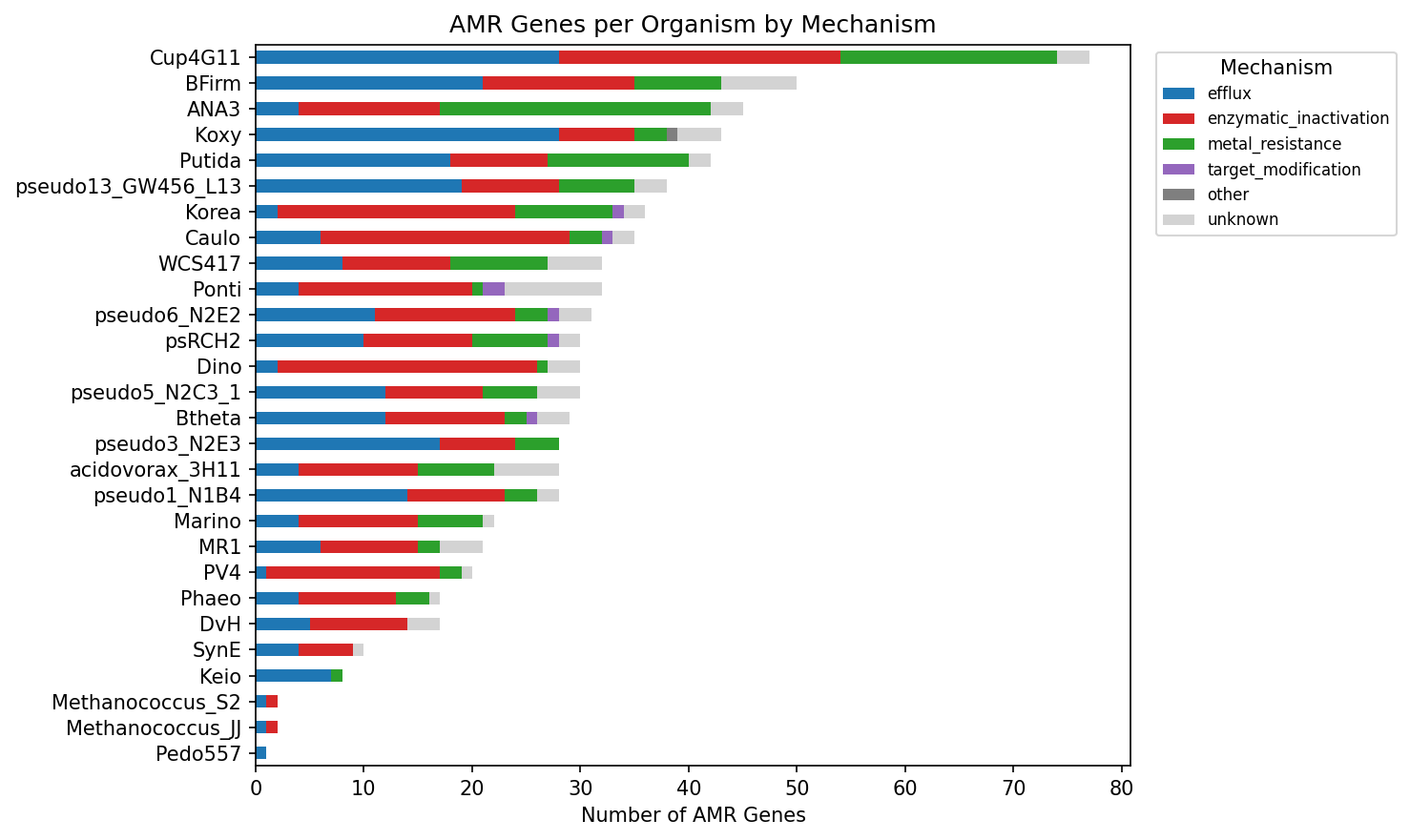
Amr Genes Per Organism
Antibiotic Validation
Class Matched Heatmap
Fitness By Mechanism Preview
Fitness Core Vs Accessory Preview
Forest Plot Amr Fitness
H2 Mechanism Stratification
H3 Core Vs Accessory
Mechanism X Conservation
Stratification Overview
Notebooks
01_data_assembly.ipynb
01 Data Assembly
View notebook →
02_fitness_cost_analysis.ipynb
02 Fitness Cost Analysis
View notebook →
03_antibiotic_validation.ipynb
03 Antibiotic Validation
View notebook →
04_stratification.ipynb
04 Stratification
View notebook →
04b_followup_analyses.ipynb
04B Followup Analyses
View notebook →