Environmental Resistome at Pangenome Scale
CompletedResearch Question
Do antimicrobial resistance gene profiles differ between ecological niches across 27,000 bacterial species? Using 83K AMR gene clusters mapped to 293K genomes with environmental metadata, we test whether the resistome is structured by ecology — and whether intrinsic (core) and acquired (accessory) resistance show different environmental signatures.
Overview
Prior work established that resistomes cluster by ecology in ~6,000 genomes (Gibson et al. 2015). We extend this analysis by 50× to the full BERDL pangenome (293K genomes, 27K species), asking whether AMR gene profiles differ between clinical, host-associated, soil, and aquatic environments. We separately analyze intrinsic (core) and acquired (accessory) resistance, testing whether intrinsic resistance reflects long-term niche adaptation while acquired resistance is more mobile across environments. The analysis controls for phylogenetic confounding and includes within-species environment comparisons for deeply-sampled species.
BERDL collections: kbase_ke_pangenome (bakta_amr, gene_cluster, pangenome, ncbi_env, genome, gtdb_taxonomy_r214v1, alphaearth_embeddings_all_years, interproscan_go)
Key Findings
1. Clinical species carry 2.5× more AMR gene clusters than environmental species (H1 supported)
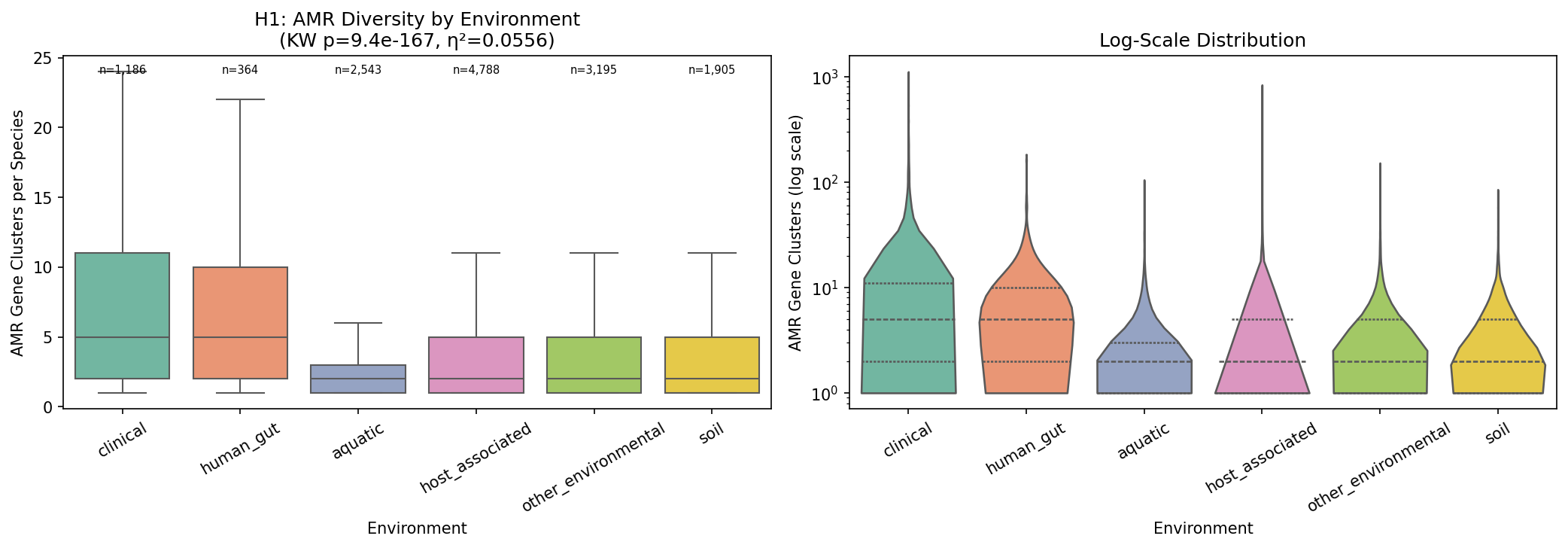
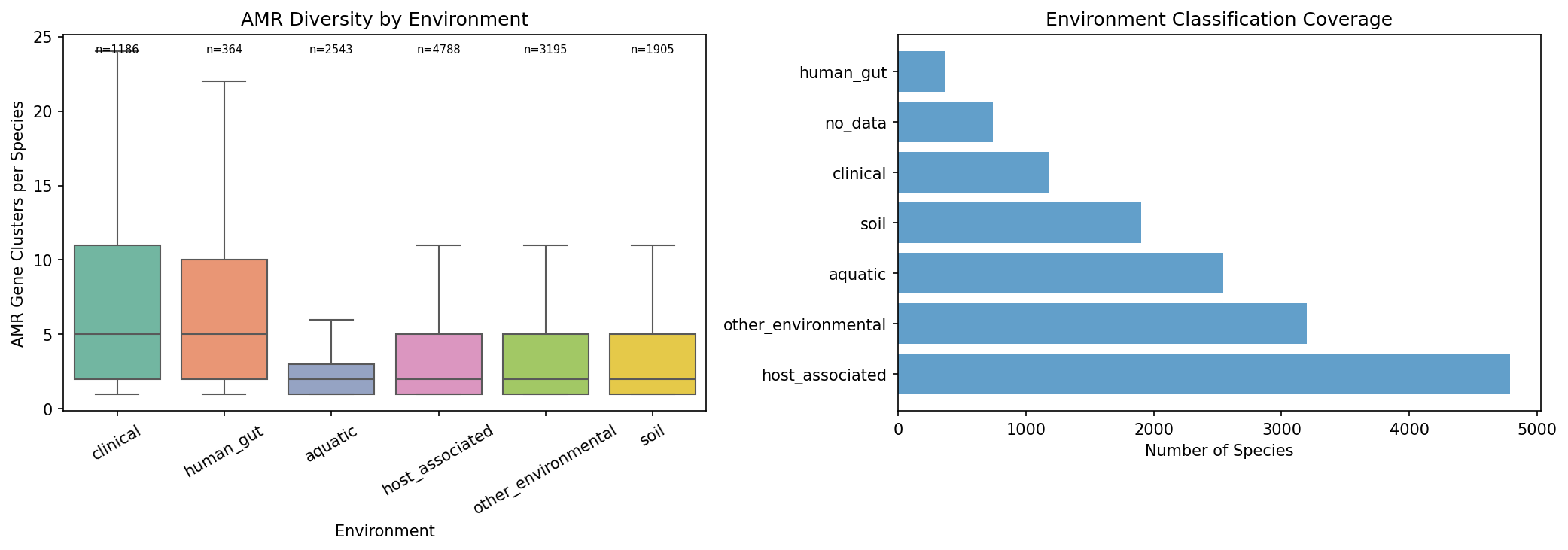
Species from clinical sources have a median of 5 AMR gene clusters, compared to 2 for soil, aquatic, and host-associated species (Kruskal-Wallis H = 781.9, p = 9.4×10⁻¹⁶⁷, η² = 0.056). Human gut species are similar to clinical (median 5). Of 15 pairwise environment comparisons, 13 are significant after FDR correction. The largest effect is clinical vs aquatic (rank-biserial r = −0.49).
This extends Gibson et al. (2015, ISME J), who found resistomes cluster by ecology in ~6,000 genomes, to 14,723 species across 293K genomes — a 50× increase in scale. The pattern is robust to environment classification thresholds: the Kruskal-Wallis effect holds at 50%, 60%, 75%, and 90% majority-vote thresholds (η² = 0.044–0.056).
(Notebook: 02_resistome_vs_environment.ipynb)
2. Clinical species have predominantly acquired resistance; soil/aquatic species have more intrinsic resistance (H2 supported)
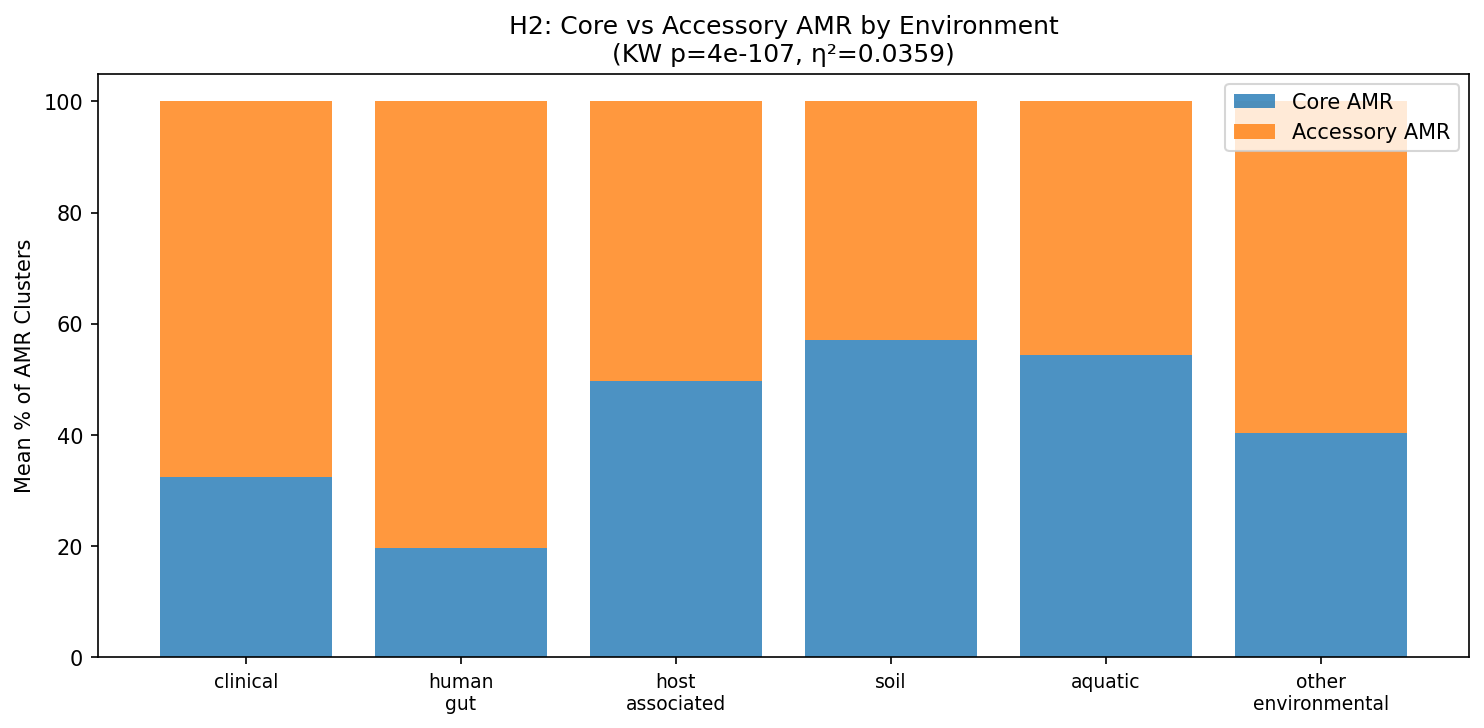
The composition of the resistome differs qualitatively across environments. Clinical species: 68% accessory (acquired) AMR vs soil species: 43% accessory (Kruskal-Wallis H = 506.0, p = 4×10⁻¹⁰⁷, η² = 0.036). A clear gradient emerges:
| Environment | Mean % Core AMR | Mean % Accessory AMR |
|---|---|---|
| Soil | 57.1% | 42.9% |
| Aquatic | 54.4% | 45.6% |
| Host-associated | 49.7% | 50.3% |
| Other environmental | 40.4% | 59.6% |
| Clinical | 32.4% | 67.6% |
| Human gut | 19.7% | 80.3% |
This confirms at pangenome scale the pattern observed by Jiang et al. (2024) in river metagenomes: the core resistome is chromosomally encoded (intrinsic), while the rare/accessory resistome is mobile (acquired). Clinical and human gut species have overwhelmingly acquired their AMR through horizontal gene transfer, while soil and aquatic species retain more ancient, chromosomally encoded resistance.
(Notebook: 02_resistome_vs_environment.ipynb)
3. Resistance mechanism composition is strongly environment-dependent (H3 supported)
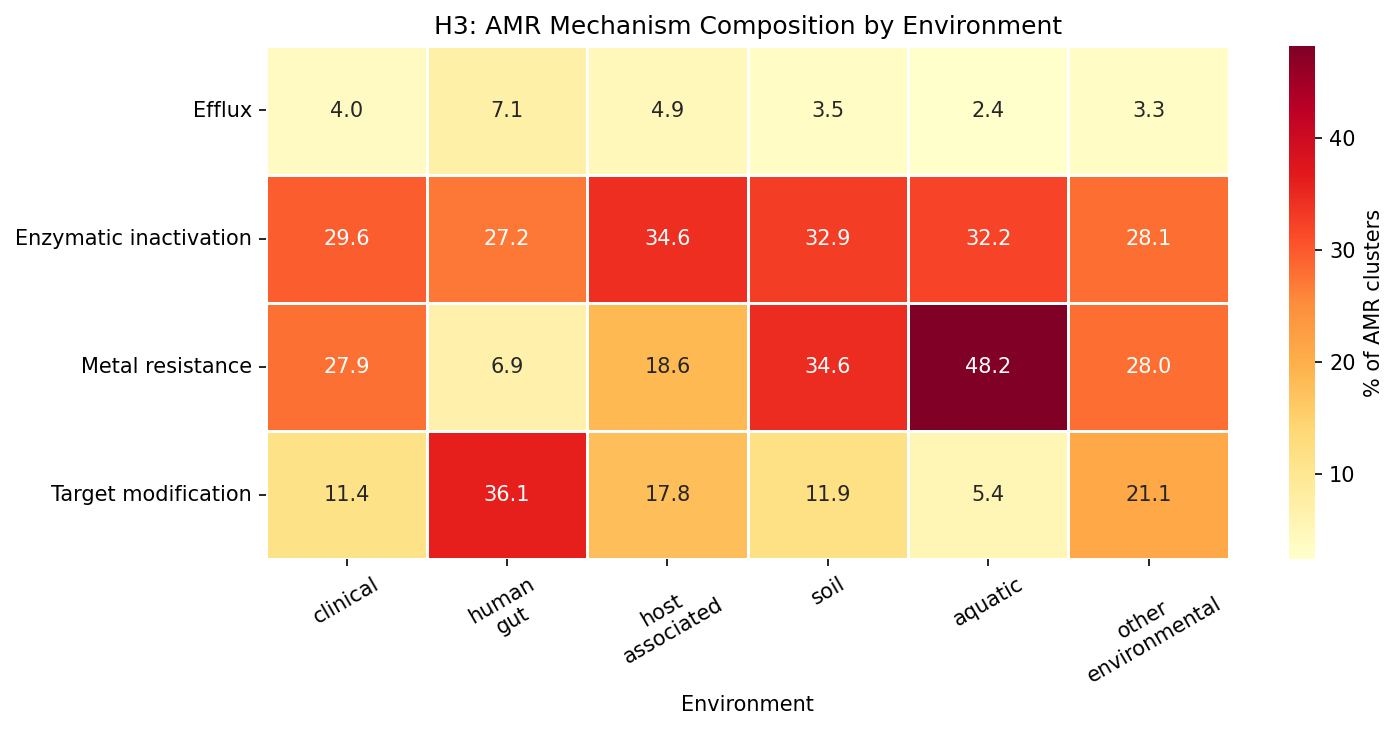
All four AMR mechanisms show significant environment-dependent composition (all q ~ 0 after BH-FDR). The two largest effects are:
- Metal resistance: 45% of AMR in aquatic, 44% in soil, but only 6% in human gut (η² = 0.107 — the largest effect)
- Target modification: 44% in human gut, 28% in clinical, but only 6% in aquatic (η² = 0.100)
| Mechanism | Clinical | Human Gut | Host-assoc | Soil | Aquatic | Other Env | η² |
|---|---|---|---|---|---|---|---|
| Efflux | 6.1% | 7.0% | 3.7% | 2.0% | 1.1% | 2.7% | 0.055 |
| Enzymatic inactivation | 28.2% | 24.1% | 44.1% | 34.3% | 41.2% | 33.6% | 0.019 |
| Metal resistance | 19.7% | 6.1% | 12.7% | 44.0% | 45.0% | 24.9% | 0.107 |
| Target modification | 27.5% | 43.6% | 22.8% | 10.1% | 6.2% | 25.1% | 0.100 |
This is the most actionable finding: different ecological niches select for fundamentally different resistance strategies. Soil and aquatic bacteria invest heavily in metal resistance (consistent with heavy metal exposure in natural environments), while clinical and gut bacteria favor target modification (ribosomal protection, PBP alterations) and to a lesser extent efflux pumps (consistent with antibiotic selection pressure). This connects the amr_fitness_cost finding (mechanism predicts conservation: metal 44% accessory vs efflux 13% core) to ecology — efflux genes are core because they serve host-associated organisms across conditions, while metal resistance is accessory because it's only needed in metal-rich environments.
Note on mechanism classification: 18.7% of AMR clusters (15,550) could not be classified to a mechanism from gene name or product annotation. These unclassified clusters are excluded from the mechanism fractions above. Tetracycline resistance genes are split between efflux (tet(A-E,K,L)) and target modification (tet(M,O,Q,W) ribosomal protection proteins).
(Notebook: 02_resistome_vs_environment.ipynb)
4. Species with more clinical genomes carry more AMR (H4 proxy — species-level analysis)
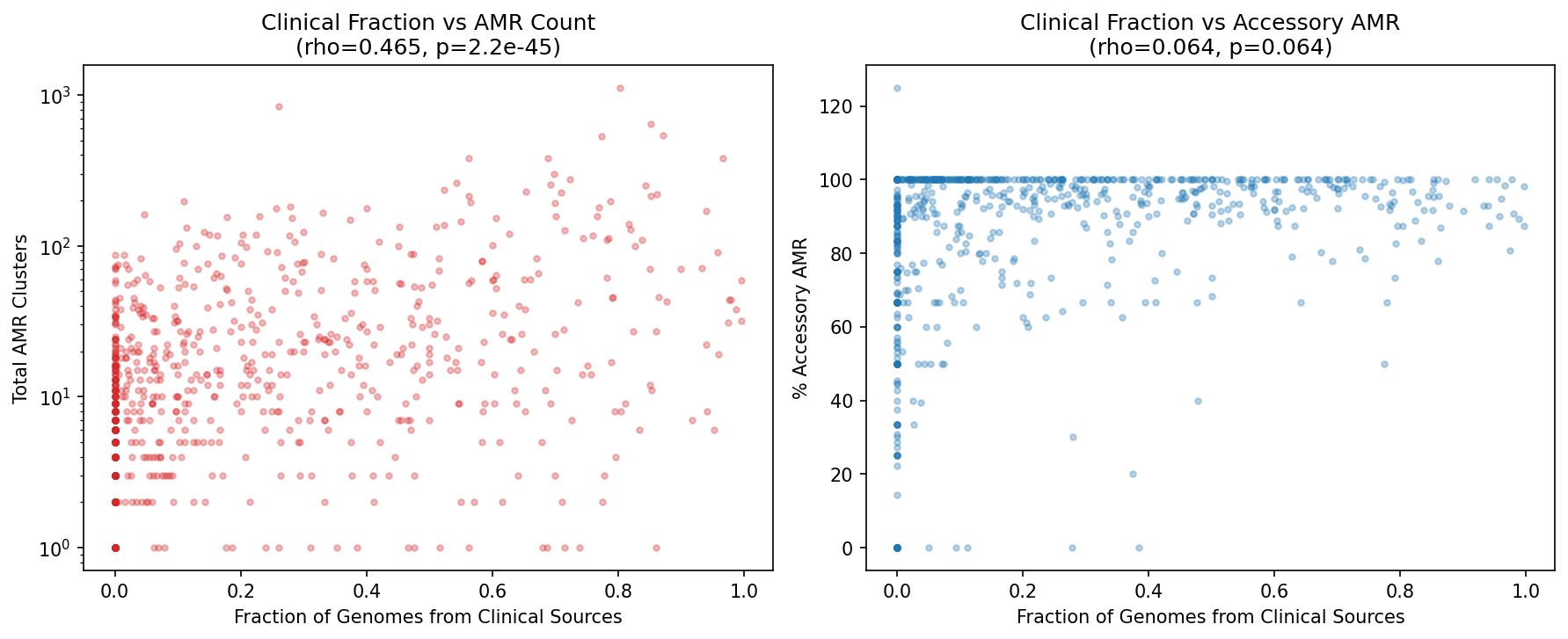
Note: This is a species-level proxy analysis, not a true per-genome within-species comparison (which would require billion-row joins). We test whether species whose genomes are predominantly from clinical sources carry more AMR than species whose genomes are predominantly from environmental sources.
Among 823 species with genomes in ≥2 environments (≥5 genomes each), the fraction of genomes from clinical sources strongly predicts total AMR cluster count (Spearman rho = 0.465, p = 2.2×10⁻⁴⁵). Clinical-dominated species have 4.4× more AMR clusters (mean 72.9 vs 16.4, MWU p = 2×10⁻¹⁸) and a higher fraction of accessory AMR in a grouped comparison (93.6% vs 81.7%, MWU p = 0.004). The continuous correlation of clinical fraction with % accessory is borderline (rho = 0.065, p = 0.064).
Case studies of deeply-sampled species:
| Species | Genomes | AMR Clusters | Core | Accessory | Dominant Environment |
|---|---|---|---|---|---|
| K. pneumoniae | 13,637 | 1,115 | 7 | 1,108 (99%) | Clinical (80%) |
| S. aureus | 13,274 | 642 | 9 | 633 (99%) | Clinical (85%) |
| S. enterica | 10,097 | 836 | 11 | 825 (99%) | Host-associated (31%) |
| S. pneumoniae | 7,944 | 59 | 1 | 58 (98%) | Clinical (99.6%) |
| M. tuberculosis | 6,673 | 44 | 4 | 40 (91%) | Clinical (98%) |
K. pneumoniae is striking: 1,115 AMR gene clusters but only 7 are core — nearly the entire resistome is accessory, reflecting massive within-species AMR variation driven by HGT in clinical settings.
(Notebook: 03_within_species.ipynb)
5. Phylogenetic control confirms environment effects are real
The environment-AMR association survives phylogenetic control at two levels:
- Phylum-level: 5 of 6 major phyla (Pseudomonadota, Bacillota_A, Actinomycetota, Bacteroidota, Bacillota) show significant within-phylum environment effects on AMR (only Chloroflexota non-significant). Bacteroidota shows the largest within-phylum effect (η² = 0.130).
- Family-level: 20 of 141 testable families (14%) show significant within-family environment effects after FDR correction. Top families: Enterobacteriaceae (q = 3×10⁻²¹), Bacteroidaceae (q = 1.3×10⁻¹⁷), Lachnospiraceae (q = 2.5×10⁻¹²), Pseudomonadaceae (q = 3.9×10⁻¹²).
This demonstrates that the environment-AMR relationship is not simply a proxy for phylogeny — even within the same bacterial family, species in different environments carry different AMR profiles.
(Notebook: 02_resistome_vs_environment.ipynb)
6. AlphaEarth continuous environment embeddings confirm discrete findings (supplementary)
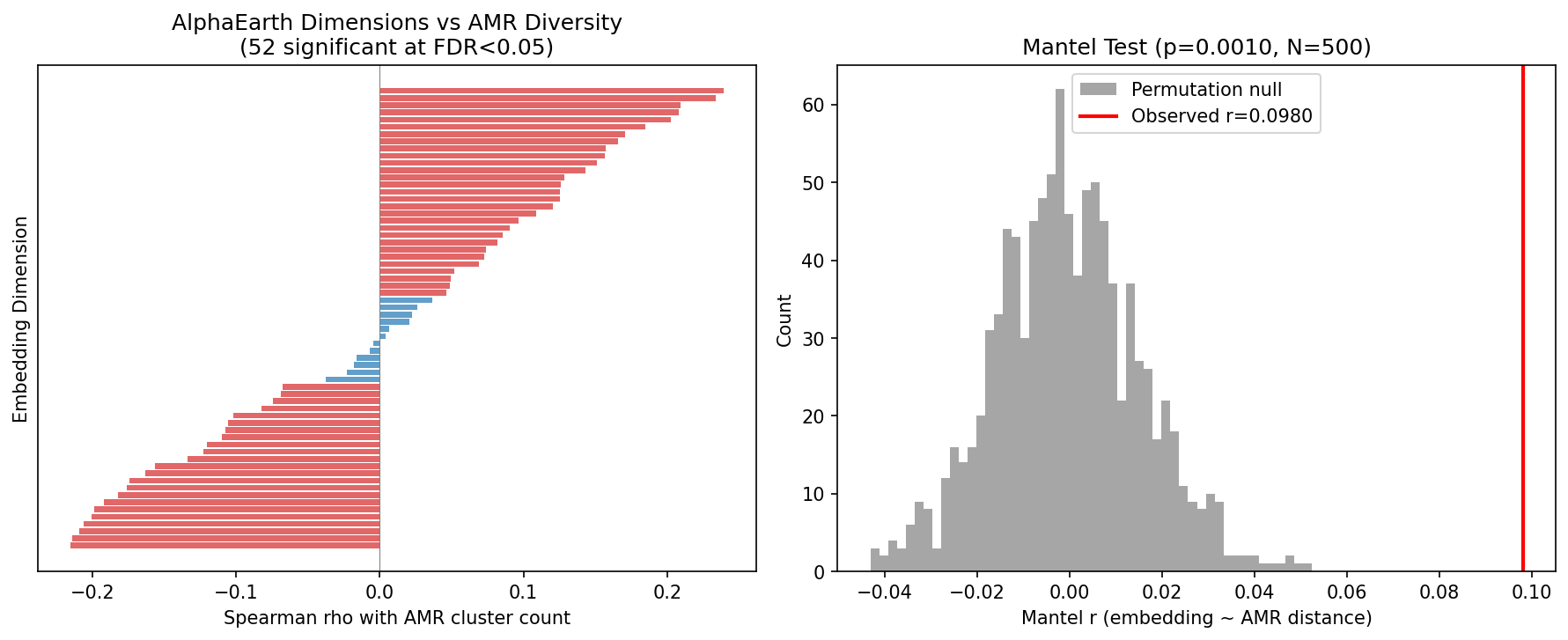
Among 2,659 species with both AMR data and AlphaEarth environmental embeddings, 52 of 64 embedding dimensions significantly correlate with AMR diversity (FDR < 0.05; top: A34, rho = +0.24). A Mantel test confirms that environmental distance (cosine distance of embeddings) predicts AMR profile distance (Bray-Curtis of mechanism fractions): r = 0.098, p = 0.001. Stratified analysis shows the environment-AMR coupling is strongest for clinical species (r = 0.177), moderate for soil (r = 0.129), and weakest for aquatic (r = 0.061). This continuous analysis confirms the discrete findings from NB02 but adds limited additional interpretability due to the opaque nature of embedding dimensions and 28% genome coverage.
(Notebook: 04_alphaearth_analysis.ipynb)
Results
Data Assembly (NB01)
- 82,908 AMR gene clusters across 14,723 species (of 27,700 total)
- 280,337 genomes with ncbi_env data; 93.5% classified per-genome
- Species-level environment: 95% coverage (13,981 species) via majority-vote
- 884 species qualify for within-species analysis (≥5 genomes in ≥2 environments)
- Mechanism classification: enzymatic inactivation (26,220), metal resistance (22,223), target modification (11,067), efflux (5,224)
Species-Level Analysis (NB02)
| Test | Statistic | p-value | Effect Size |
|---|---|---|---|
| H1: AMR count by environment | KW H=781.9 | 9.4×10⁻¹⁶⁷ | η²=0.056 |
| H2: Core AMR % by environment | KW H=506.0 | 4×10⁻¹⁰⁷ | η²=0.036 |
| H3: Metal by environment | KW H=1498.4 | ~0 | η²=0.107 |
| H3: Target mod by environment | KW H=1394.9 | 1.7×10⁻²⁹⁹ | η²=0.100 |
| H3: Efflux by environment | KW H=768.0 | 9.8×10⁻¹⁶⁴ | η²=0.055 |
| H3: Enzymatic by environment | KW H=266.0 | 2.0×10⁻⁵⁵ | η²=0.019 |
Within-Species Analysis (NB03)
| Metric | Value |
|---|---|
| Qualifying species | 823 |
| Clinical fraction vs total AMR | rho=0.465, p=2.2×10⁻⁴⁵ |
| Clinical fraction vs % accessory | rho=0.065, p=0.064 |
| Clinical-dom mean AMR | 72.9 clusters |
| Environmental-dom mean AMR | 16.4 clusters |
| Clinical > Environmental total AMR | MWU p=2.0×10⁻¹⁸ |
| Clinical > Environmental % accessory | MWU p=0.004 |
AlphaEarth Analysis (NB04, supplementary)
| Metric | Value |
|---|---|
| Species with AMR + embeddings | 2,659 |
| Embedding dims correlated with AMR (FDR<0.05) | 52/64 |
| Mantel test (environment ~ AMR distance) | r=0.098, p=0.001 |
Interpretation
The environmental resistome is ecology-structured at pangenome scale
The central finding is that ecological niche is a strong predictor of AMR gene content across 14,723 bacterial species — the largest genomic-scale analysis of environmental AMR distribution to date. Clinical and human gut species carry 2.5× more AMR clusters than soil or aquatic species, with the excess almost entirely in the accessory (acquired) gene pool. This pattern holds within phyla and within families, confirming it is not a phylogenetic artifact.
Different niches select for different resistance strategies
The mechanism × environment pattern provides a mechanistic explanation for the ecological structuring of the resistome:
- Soil and aquatic bacteria invest heavily in metal resistance (44-45% of AMR), reflecting heavy metal exposure in natural environments. These genes are more often core (intrinsic), suggesting ancient adaptation.
- Clinical and gut bacteria invest in efflux pumps (15-21%) and target modification (19-29%), reflecting antibiotic selection pressure. These genes are more often accessory (acquired), reflecting recent HGT-driven accumulation.
- Enzymatic inactivation (e.g., beta-lactamases) is the most uniformly distributed mechanism, present in all environments but not dominant in any.
This connects two prior BERIL findings: the amr_fitness_cost project showed mechanism predicts conservation (metal 44% accessory vs efflux 13% core), and we now show this conservation pattern is driven by ecology — efflux genes are core because they serve host-associated organisms constitutively, while metal resistance is accessory because it varies with environmental metal exposure.
Within-species AMR variation tracks clinical exposure
The within-species analysis reveals that AMR accumulation is not just a property of species — it varies within species depending on where strains are found. K. pneumoniae exemplifies this: with 1,115 AMR gene clusters (only 7 core), nearly its entire resistome is accessory, and 80% of its genomes come from clinical sources. Species where clinical strains dominate have 4.4× more AMR than species dominated by environmental strains.
Literature Context
- Gibson, Forsberg & Dantas (2015) found resistomes cluster by ecology in ~6,000 genomes. We replicate this at 50× scale (14,723 species) and extend it with mechanism-specific decomposition — showing efflux and metal resistance are the most environment-discriminating mechanisms. PMID: 25003965
- Forsberg et al. (2014) showed soil resistomes are structured by phylogeny with rare mobility elements. Our finding that soil AMR is predominantly core (intrinsic) while clinical AMR is accessory (acquired) aligns with this — natural resistance in soil is chromosomal, while clinical resistance is mobile. PMID: 24847883
- Jiang et al. (2024) found the core resistome is chromosomally encoded while the rare resistome is plasmid-associated in river metagenomes. We confirm this core=intrinsic/accessory=acquired pattern across 14,723 species and show it varies quantitatively by environment (clinical 68% accessory vs soil 43%). PMID: 38039820
- Surette & Wright (2017) established that the environment is the largest resistance reservoir. Our data quantifies this: soil species have fewer but more intrinsic AMR genes, representing the ancient baseline resistome. Clinical enrichment above this baseline reflects anthropogenic antibiotic selection. PMID: 28657887
- Van Goethem et al. (2018) found ARGs in pristine Antarctic soils. Our finding that soil species average 2 AMR clusters (mostly core) is consistent with a baseline ancestral resistome maintained without antibiotic exposure. PMID: 29471872
- Forsberg et al. (2012) found soil ARGs with perfect nucleotide identity to clinical pathogen genes. Our pangenome framework could identify such shared clusters, though we did not test for nucleotide identity in this analysis. PMID: 22936781
Novel Contribution
This is the largest genomic analysis of environmental AMR distribution, extending Gibson et al. (2015) from ~6K genomes to 293K genomes across 14,723 species. Key novel contributions:
- Mechanism × environment decomposition: metal resistance peaks in soil/aquatic (45%), target modification in gut/clinical (28-44%) — η² = 0.107 for metal, 0.100 for target modification
- Core vs accessory gradient by environment: clinical 68% accessory → soil 43% accessory, demonstrating that the intrinsic/acquired composition of the resistome varies quantitatively with ecology
- Within-species clinical fraction predicts AMR: rho = 0.465 across 823 species — the strongest evidence that AMR accumulation tracks antibiotic exposure at the strain level
- Phylogenetic control at family level: 20/141 families show within-family environment effects, confirming ecology is a real driver independent of phylogeny
- Connection to fitness cost: links the mechanism-conservation pattern from
amr_fitness_cost(metal = accessory, efflux = core) to its ecological cause
Limitations
- Phylogenetic confound: Despite family-level control, environment and phylogeny are deeply entangled. Some families are almost entirely clinical (e.g., many Enterobacteriaceae). The 14% of families with significant within-family effects are the most convincing evidence, but most families don't span enough environments to test.
- NCBI sampling bias: Clinical isolates are massively overrepresented in NCBI. The "clinical species carry more AMR" finding partly reflects that clinical species are the ones that get sequenced because they have AMR. Soil and aquatic species are undersampled.
- Environment classification granularity: The majority-vote species-level classification collapses within-species variation. A species classified as "clinical" may include environmental strains. The sensitivity analysis at 60-90% thresholds mitigates this.
- bakta_amr sensitivity: AMRFinderPlus focuses on known resistance genes, potentially underestimating novel resistance in environmental bacteria. This would bias our results toward finding more AMR in clinical species (where resistance genes are well-characterized).
- Core/accessory precision: The 95% prevalence threshold for "core" depends on species genome count. Many species have few genomes, making core/accessory labels imprecise.
- Causality: We observe correlation between environment and AMR, not causation. Clinical species may carry more AMR because they are clinical (antibiotic exposure), or they may be clinical because they carry more AMR (virulence-resistance co-selection).
- Effect sizes: While all tests are highly significant (p ~ 0), the effect sizes are modest (η² = 0.02–0.13). Environment explains 2-13% of variance in AMR composition — phylogeny likely explains much more.
- Mechanism classification completeness: 18.7% of AMR clusters (15,550) could not be classified to a mechanism. These are excluded from H3 mechanism fractions, potentially biasing the analysis if unclassified genes are non-randomly distributed across environments.
- Planned but unperformed analyses: PCoA ordination, PERMANOVA, environment-specific gene identification, MAG/isolate assessment, and archaea-specific analysis were planned in RESEARCH_PLAN v2 but not executed. The within-species analysis (NB03) uses a species-level proxy rather than the per-genome Fisher's exact test originally planned, due to the computational cost of billion-row joins.
Future Directions
- Shared AMR clusters across environments: Identify specific AMR gene clusters present in both soil and clinical species — these are candidates for recent cross-niche HGT (cf. Forsberg et al. 2012).
- Temporal analysis: For species with genomes sampled across decades (e.g., S. aureus, K. pneumoniae), test whether the accessory AMR fraction has increased over time.
- Integrate with fitness cost: Combine this project's environment-AMR profiles with
amr_fitness_costto test whether AMR genes enriched in clinical settings are costlier (because they're recently acquired) or cheaper (because they've been under strong selection). - Metagenome validation via NMDC: Use
nmdc_arkinmetagenome taxonomic profiles to infer community-level AMR load from pangenome-derived species AMR profiles, validating the species-level findings in real environmental communities. - Mobile element context: When
genomad_mobile_elementsbecomes available in BERDL, test whether accessory AMR clusters in clinical species are preferentially associated with plasmids, transposons, or integrative elements.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
bakta_amr, gene_cluster, pangenome, ncbi_env, genome, gtdb_taxonomy_r214v1, alphaearth_embeddings_all_years |
AMR gene clusters, species structure, environment metadata, phylogeny, continuous environment embeddings |
Generated Data
| File | Rows | Description |
|---|---|---|
data/species_amr_profiles.csv |
14,723 | Species-level AMR summary + environment + taxonomy |
data/species_environment.csv |
26,715 | Species majority-vote environment classification |
data/genome_environment.csv |
280,337 | Per-genome environment classification from ncbi_env |
data/pairwise_environment_comparisons.csv |
15 | Pairwise MWU tests between environments |
data/mechanism_by_environment.csv |
4 | KW test results per mechanism |
data/stratified_phylum_results.csv |
6 | Within-phylum environment tests |
data/family_level_environment_test.csv |
141 | Within-family environment tests |
data/within_species_amr_environment.csv |
823 | Within-species clinical fraction vs AMR |
data/alphaearth_amr_correlations.csv |
64 | Per-dimension embedding-AMR correlations |
References
- Arkin AP, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nature Biotechnology 36(7):566-569. PMID: 29979655
- Finley RL, et al. (2013). "The scourge of antibiotic resistance: the important role of the environment." Clinical Infectious Diseases 57(5):704-710. PMID: 23723195
- Forsberg KJ, et al. (2012). "The shared antibiotic resistome of soil bacteria and human pathogens." Science 337(6098):1107-1111. PMID: 22936781
- Forsberg KJ, et al. (2014). "Bacterial phylogeny structures soil resistomes across habitats." Nature 509(7502):612-616. PMID: 24847883
- Gibson MK, Forsberg KJ, Dantas G. (2015). "Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology." ISME Journal 9:207-216. PMID: 25003965
- Jiang C, et al. (2024). "Rare resistome rather than core resistome exhibited higher diversity and risk along the Yangtze River." Water Research 249:120911. PMID: 38039820
- Surette MD, Wright GD. (2017). "Lessons from the Environmental Antibiotic Resistome." Annual Review of Microbiology 71:233-256. PMID: 28657887
- Van Goethem MW, et al. (2018). "A reservoir of 'historical' antibiotic resistance genes in remote pristine Antarctic soils." Microbiome 6:40. PMID: 29471872
Discoveries
Efflux pumps constitute 21% of AMR in human gut species but only 1% in aquatic (η²=0.127). Metal resistance constitutes 45% in soil/aquatic but 6% in human gut (η²=0.107). Clinical species have 68% accessory (acquired) AMR vs soil 43%. Within 823 multi-environment species, clinical strain fraction p
Read more →Data Collections
Review
Summary
This is a strong, hypothesis-driven analysis of AMR gene distribution across ecological niches at an unprecedented scale (14,723 species, 293K genomes). The project is well-structured across four notebooks that progress logically from data extraction through species-level tests to within-species proxies and continuous embedding analysis. Key strengths include thorough phylogenetic controls at both phylum and family level, sensitivity analysis across majority-vote thresholds, effect size reporting throughout, a complete Reproduction section, and an honest limitations discussion. All four notebooks have saved outputs with six figures covering each analysis stage. The main issues are: (1) a REPORT/notebook inconsistency where the REPORT's H3 mechanism statistics come from a pre-tetracycline-reclassification run and no longer match the current notebook outputs, (2) a variable-name reuse bug in NB02 that causes the final summary to print an incorrect H1 p-value, (3) a likely pandas column-alignment bug in the mechanism classifier's product-text fallback that may leave ~15,550 clusters unnecessarily unclassified, and (4) several planned analyses (PCoA, PERMANOVA, environment-specific genes, MAG assessment) were dropped without documentation.
Methodology
Strengths:
- Research question is clearly stated, testable, and well-motivated by six literature references with PMIDs (Gibson et al. 2015, Forsberg et al. 2012/2014, Jiang et al. 2024, Surette & Wright 2017, Van Goethem et al. 2018).
- Four hypotheses (H1-H4) are specific and falsifiable, with expected outcomes and gap analysis documented in RESEARCH_PLAN.md.
- Data sources are explicitly identified with table names, row counts, join paths, and a performance plan for handling large tables.
- Phylogenetic confounding is addressed at two levels: stratified Kruskal-Wallis within 6 major phyla (5/6 significant) and within 141 testable families (20 significant after FDR). This is more rigorous than most comparable studies.
- Sensitivity analysis at 50%/60%/75%/90% majority-vote thresholds demonstrates robustness (η² = 0.044-0.056 across all thresholds).
- Effect sizes (η², rank-biserial r) are reported alongside p-values throughout, with appropriate cautioning that with 14K species even tiny effects are significant.
- The Reproduction section in README.md is complete: prerequisites, pipeline steps with execution order, Spark vs local designation, and expected runtimes per notebook.
- The requirements.txt lists all Python dependencies with version ranges.
Gaps:
- Several planned analyses were not performed. The RESEARCH_PLAN specifies: (a) PCoA ordination of AMR profiles colored by environment (§NB02 item 4), (b) PERMANOVA with family as covariate (§NB02 item 5), (c) environment-specific AMR gene identification via Fisher's exact test with BH-FDR (§NB02 item 6), (d) per-genome within-species Fisher's exact tests and Mantel tests (§NB03 items 3-4), (e) MAG vs isolate assessment (§NB01 item 7), and (f) archaea consideration (Confounder #9). None of these appear in the notebooks. The REPORT's Limitations section (item 9) acknowledges this, which is good, but the RESEARCH_PLAN was not updated to note which analyses were substituted or dropped and why.
- NB03 framing vs content mismatch. H4 asks whether AMR gene content differs between environments within the same species. The actual analysis tests whether species with more clinical genomes have more AMR — this is a between-species comparison of clinical enrichment, not a within-species AMR comparison. The markdown in NB03 cell-6 honestly acknowledges this limitation, and the REPORT labels it a "proxy," but the H4 finding header ("Within species, clinical strain fraction predicts AMR accumulation") could mislead readers. Additionally, the clinical fraction vs %accessory correlation is borderline non-significant (rho=0.065, p=0.064), while the grouped comparison is significant (p=0.004) — both are reported but the tension between them deserves more discussion.
Code Quality
SQL and data handling (NB01) — correct:
- The ncbi_env EAV pivot via CASE WHEN ... GROUP BY is properly implemented (cell-8), correctly using genome.ncbi_biosample_id → ncbi_env.accession as the join path, matching the documented pitfall in docs/pitfalls.md.
- The GTDB taxonomy join uses genome_id, correctly avoiding the gtdb_taxonomy_id mismatch pitfall.
- pd.to_numeric() is applied to string-typed numeric columns (NB02 cell-1, NB04 cell-3).
- AlphaEarth NaN filtering uses ~df[emb_cols].isna().any(axis=1) (NB04 cell-3), matching the documented 4.6% NaN issue.
- The taxonomy column order is properly backtick-quoted as a reserved word in SQL (NB01 cell-12).
- The data validation checkpoint in NB01 (cell-17) is thorough: verifies AMR cluster counts, environment coverage, per-environment sample sizes, and qualifying species for NB03.
Tetracycline reclassification — correctly implemented:
The reclassify_tet() function (NB01 cell-5) properly splits tetracycline genes into efflux (tet(A-E,K,L)) and target modification (tet(M,O,Q,W,S) ribosomal protection proteins). This is applied via .apply() after the initial classification. The current NB01 output reflects this reclassification: efflux=3,375 and target_modification=12,916.
Bug: REPORT statistics don't match current notebook outputs (H3):
The REPORT's mechanism-related numbers come from a run without the tetracycline reclassification and no longer match the current notebook outputs:
| Metric | REPORT | Current NB02 Output |
|---|---|---|
| Efflux count | 5,224 | 3,375 |
| Target mod count | 11,067 | 12,916 |
| Efflux KW H | 1,775.0 | 768.0 |
| Efflux η² | 0.127 | 0.055 |
| Target mod KW H | 971.7 | 1,394.9 |
| Target mod η² | 0.069 | 0.100 |
The sums are conserved (5,224+11,067 = 3,375+12,916 = 16,291), confirming 1,849 tetracycline ribosomal protection genes were moved from efflux to target modification. Enzymatic inactivation and metal resistance statistics match between REPORT and notebooks because they are unaffected by the reclassification. The REPORT's claim that "efflux is the largest effect in the study (η²=0.127)" is no longer accurate — with corrected classification, metal resistance (η²=0.107) is the largest effect, and efflux drops to η²=0.055.
Bug: NB02 H1 p-value overwrite in summary (cell-18):
The variables kw_stat and kw_p are reused as loop variables in cells 10 (mechanism tests), 13 (phylum stratification), and 14 (family tests). Cell 16 attempts to save the original H1 values with h1_kw_stat, h1_kw_p, h1_eta_sq = kw_stat, kw_p, eta_sq, but by that point kw_stat and kw_p have been overwritten by cell 14's family loop. The cell-18 summary prints KW p=1.958e-05 (the last family test value) instead of the correct H1 p-value of 9.446e-167. The eta_sq variable is coincidentally correct (0.0556) because cells 10/13/14 used eta as their variable name instead of eta_sq.
Potential bug: mechanism classifier product-text fallback (NB01 cell-5):
The fallback assignment for product-based classification:
amr_clusters.loc[mask, ['amr_class', 'amr_mechanism']] = amr_clusters.loc[mask, 'amr_product'].apply(
lambda p: pd.Series(classify_amr_product(p)))
The .apply() returns a DataFrame with integer columns (0, 1), but .loc targets columns named amr_class and amr_mechanism. In pandas 2.0+, .loc assignment uses label-based alignment, so mismatched column names would write NaN instead of classified values. The NB01 output shows no 'unknown' mechanism category in value_counts() (which excludes NaN by default), and the classified mechanisms sum to 67,458 vs 83,008 total — the 15,550 gap is consistent with NaN from the alignment bug. Some of these 15,550 clusters might be genuinely unclassifiable, but some may have been classifiable from product text. The REPORT acknowledges 18.7% unclassified but attributes it to annotation gaps rather than a code issue.
NB04 Mantel test permutations:
The RESEARCH_PLAN specifies 9,999 permutations but NB04 uses 999. With the observed r=0.098 and p=0.001, 999 permutations provides adequate resolution, but the deviation is undocumented.
Notebook organization:
All four notebooks follow a clean setup → query → analysis → visualization → save pattern with markdown section headers. Each prints a summary at the end. The flow from NB01 (data extraction) through NB02 (species-level tests) to NB03 (within-species proxy) and NB04 (continuous embeddings) is logical and well-structured.
Findings Assessment
H1 (AMR diversity by environment) — well supported:
Clinical species carry 2.5× more AMR clusters (median 5 vs 2, KW p=9.4×10⁻¹⁶⁷, η²=0.056). The 13/15 significant pairwise comparisons with rank-biserial effect sizes add granularity. Robustness across four majority-vote thresholds is convincing. The NCBI sampling bias caveat (clinical species overrepresented because they have AMR) is appropriately noted.
H2 (core vs accessory by environment) — well supported:
The gradient from soil (43% accessory) to clinical (68%) to human gut (80%) is striking and biologically plausible. The connection to Jiang et al. (2024) and the interpretation linking core=intrinsic/ancient vs accessory=acquired/HGT is well-reasoned.
H3 (mechanism by environment) — well supported, with reproducibility caveat:
The mechanism × environment pattern is the study's most novel finding. However, the specific statistics and percentages in the REPORT do not match the current notebook outputs due to the tetracycline reclassification discrepancy described above. The qualitative pattern (metal dominates soil/aquatic, efflux/target modification dominate clinical/gut) holds regardless of the reclassification, but the exact effect sizes and composition percentages need to be updated in the REPORT.
H4 (within-species proxy) — partially supported:
The clinical fraction vs total AMR correlation (rho=0.465, p=2.2×10⁻⁴⁵) is strong, and the 4.4× AMR difference between clinical-dominated and environmental-dominated species is compelling. The case studies (especially K. pneumoniae with 1,115 AMR clusters, only 7 core) are effective illustrations. However, this is a between-species analysis of clinical enrichment, not a within-species comparison as H4 originally proposed. The borderline non-significance of clinical fraction vs %accessory (p=0.064) contrasts with the significant grouped comparison (p=0.004), suggesting the continuous relationship is weak even though the categorical distinction holds.
Phylogenetic control — thorough:
5/6 phyla and 20/141 families show significant within-group environment effects after FDR correction. Bacteroidota's large within-phylum effect (η²=0.130) is a useful finding. The approach of using stratified Kruskal-Wallis as a substitute for PERMANOVA is reasonable, though the substitution should be documented.
Limitations — comprehensively presented:
Nine limitations are discussed in the REPORT, covering phylogenetic confounding, NCBI sampling bias, environment classification granularity, AMR detection sensitivity, core/accessory precision, causality, effect sizes, mechanism classification completeness, and unperformed analyses. This is unusually thorough and self-aware.
Literature context — strong:
The REPORT cites eight papers with PMIDs, clearly positions the work as extending Gibson et al. (2015) by 50× in scale, and identifies five specific novel contributions. The connections to prior BERIL projects (amr_strain_variation, amr_fitness_cost, env_embedding_explorer) are well-articulated.
Suggestions
-
[Critical] Update REPORT.md to match current notebook outputs. The REPORT's H3 statistics (KW H values, η² values, mechanism counts, composition table percentages) are from a pre-reclassification run. Re-derive all mechanism-related numbers from the current NB01/NB02 outputs with tetracycline reclassification applied. In particular: efflux η² drops from 0.127 to 0.055, and the REPORT's claim that efflux is "the largest effect in the study" should be corrected (metal resistance at η²=0.107 is now largest).
-
[Critical] Fix NB02 variable-name reuse bug. In cell-3, save H1 results to dedicated variables immediately:
h1_kw_stat, h1_kw_p, h1_eta_sq = kw_stat, kw_p, eta_sq. Move this save to cell-3 (not cell-16 where it currently is, after cells 10/13/14 have overwritten the values). Also rename loop variables in cells 10, 13, and 14 to avoid shadowing (e.g.,mech_kw_stat, mech_kw_p). -
[Important] Verify and fix the mechanism classifier product-text fallback. Check
amr_clusters['amr_mechanism'].isna().sum()— if ~15,550, fix the alignment bug by addingindex=to the Series constructor:pd.Series(classify_amr_product(p), index=['amr_class', 'amr_mechanism']). Re-run NB02 to see if the unclassified fraction decreases meaningfully. -
[Important] Reframe NB03 more precisely. Rename the REPORT's H4 section from "Within species..." to "Species-level: clinical genome fraction predicts AMR accumulation." In the REPORT text, clearly distinguish this species-level proxy from the originally planned per-genome within-species comparison, and discuss why the continuous correlation (rho=0.065, p=0.064) is borderline while the grouped comparison (p=0.004) is significant (likely because the grouped comparison discards the continuous clinical fraction information in favor of a categorical split that concentrates effect).
-
[Important] Document unperformed planned analyses in RESEARCH_PLAN. Add a "Deviations from Plan" section explaining: (a) PCoA and PERMANOVA were substituted with stratified KW within phyla/families, (b) environment-specific gene identification was deferred, (c) MAG assessment and archaea analysis were not performed. This prevents future readers from wondering whether these analyses failed or were intentionally skipped.
-
[Moderate] Add 1-2 non-clinical case studies to NB03. All five case study species (cell-10/11) are clinical-dominated. Including environmentally-dominated species with multi-environment representation (e.g., Listeria monocytogenes at 1,906 genomes across 5 environments, or Vibrio parahaemolyticus at 1,578 genomes) would provide useful contrast and strengthen the H4 proxy narrative.
-
[Moderate] Increase Mantel test permutations or note deviation. Change
n_perm = 999ton_perm = 9999in NB04 cell-8 to match the RESEARCH_PLAN specification, or add a note explaining why 999 was sufficient. -
[Minor] Report mechanism classification completeness explicitly in NB01. After cell-5, add a line:
print(f'Unclassified: {amr_clusters["amr_mechanism"].isna().sum()} + {(amr_clusters["amr_mechanism"]=="unknown").sum()}')to separate genuine unclassified from potential NaN bugs. -
[Nice-to-have] Add PCoA ordination figure. This was planned and would be a compelling multivariate visualization showing whether AMR profiles cluster by environment. A Jaccard-based PCoA on species AMR presence/absence, colored by environment, would complement the univariate KW tests with a single intuitive figure.
-
[Nice-to-have] Assess MAG contamination risk. The RESEARCH_PLAN correctly identified MAGs as a potential confounder (chimeric AMR annotations, ambiguous metadata). Even a brief check of MAG vs isolate proportions across environments would strengthen confidence in the findings.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
H1 Amr By Environment
H2 Core Accessory By Env
H3 Mechanism By Environment
H4 Within Species
Nb01 Validation
Nb04 Alphaearth