AMR Co-Fitness Support Networks
CompletedResearch Question
What genes are co-regulated with antimicrobial resistance (AMR) genes across growth conditions, and do these "support networks" explain the uniform fitness cost of resistance? Using cofitness data and ICA fitness modules from 25 bacteria, we identify the functional context in which AMR genes operate.
Overview
The amr_fitness_cost project established that AMR genes impose a universal +0.086 fitness cost that is mechanism-independent. This follow-up asks why the cost is uniform: are AMR genes embedded in larger co-regulated networks whose functional composition explains the cost? We use pairwise cofitness correlations (13.6M pairs) and ICA-derived fitness modules (1,116 modules) to map the "support network" around each AMR gene — the genes whose fitness phenotypes track with resistance across hundreds of experimental conditions.
BERDL collections: kescience_fitnessbrowser (genefitness, gene), kbase_ke_pangenome (bakta_amr, bakta_annotations, interproscan_go, interproscan_domains)
Key Findings
1. AMR genes are embedded in larger-than-average co-regulated modules
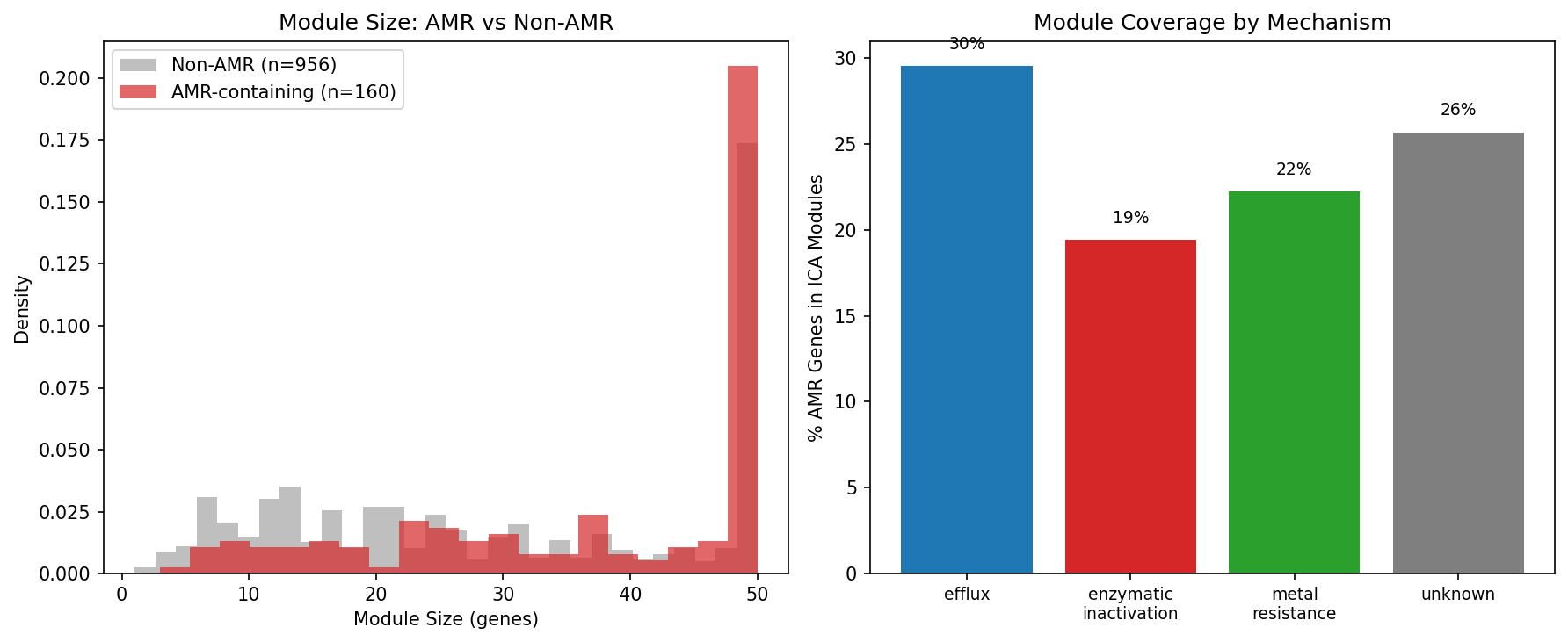
Only 24% of AMR genes (192/801) are assigned to ICA fitness modules, but the modules they inhabit are significantly larger than non-AMR modules: median 46 vs 27 genes (MWU p = 1.7×10⁻⁸). This indicates that when AMR genes are tightly co-regulated with other genes, it is within large, multi-function cellular programs, not small isolated modules. Notably, 99% (208/209) of AMR gene-module assignments are in cross-organism conserved module families, indicating these are ancient regulatory relationships. Module size does not differ between AMR mechanisms (efflux = enzymatic = 48, MWU p = 0.91).
(Notebook: 02_amr_in_modules.ipynb)
2. AMR support networks are enriched for flagellar motility and amino acid biosynthesis (H1 supported)
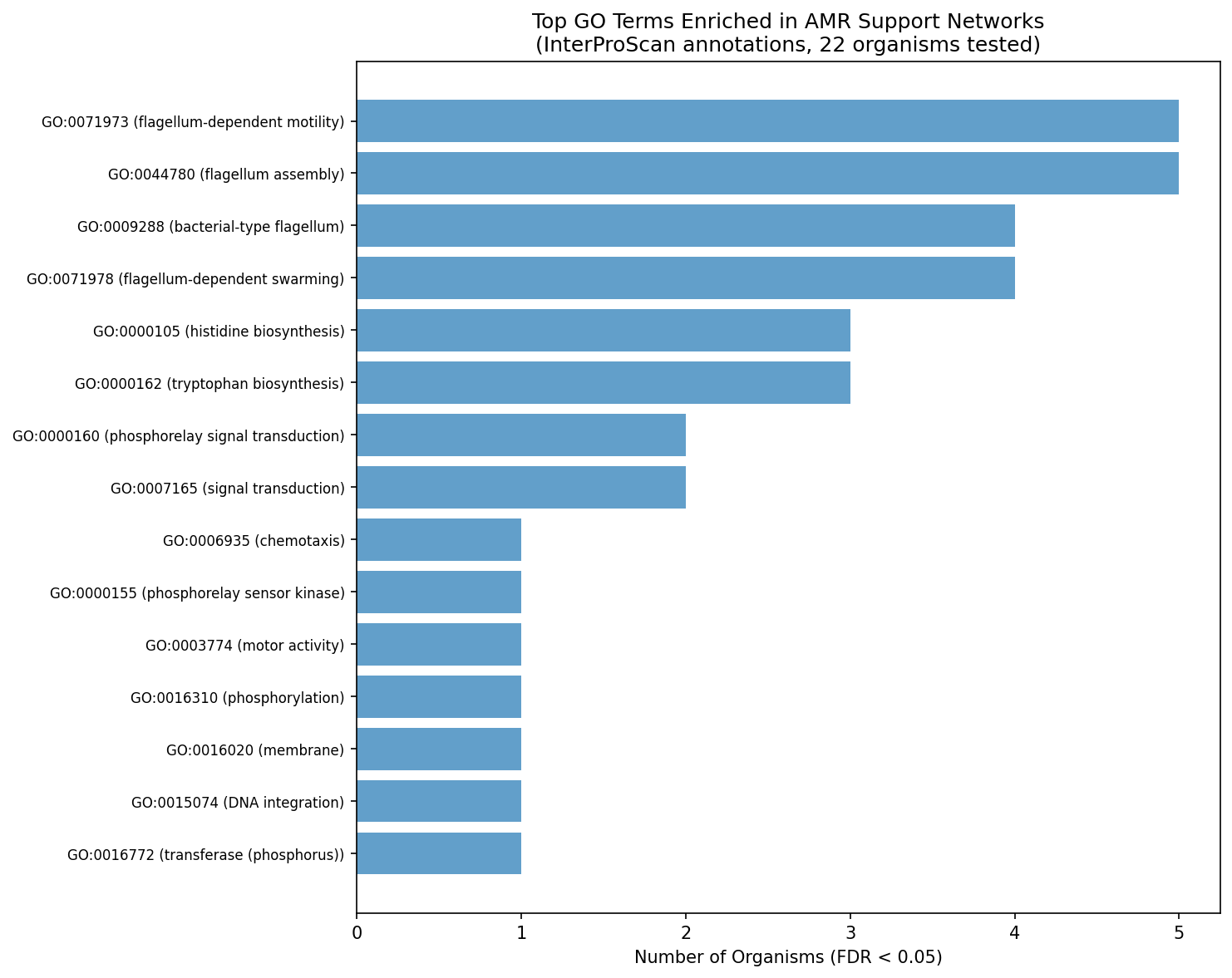
Using InterProScan GO annotations (68% gene coverage — 3.6× better than old SEED annotations), we detect significant functional enrichment in AMR cofitness neighborhoods. The top 6 GO terms enriched in ≥3 organisms (FDR < 0.05) are:
| GO Term | Description | Organisms (FDR<0.05) | Mean OR |
|---|---|---|---|
| GO:0071973 | Flagellum-dependent cell motility | 5 | 4.7 |
| GO:0044780 | Flagellum assembly | 5 | 5.3 |
| GO:0009288 | Bacterial-type flagellum | 4 | 4.9 |
| GO:0071978 | Flagellum-dependent swarming | 4 | 5.0 |
| GO:0000105 | Histidine biosynthesis | 3 | 5.3 |
| GO:0000162 | Tryptophan biosynthesis | 3 | 5.3 |
This enrichment was undetectable with old SEED annotations (0/280 significant) — a dramatic demonstration that annotation quality matters for functional genomics.
Important caveat: This enrichment may reflect shared dispensability under lab conditions rather than genuine co-regulation. FB experiments use shaken liquid culture (where flagella are useless) and often supplemented media (where biosynthesis is redundant). AMR genes, flagellar genes, and biosynthesis genes are all metabolic burdens under these conditions. A fitness-matched permutation (drawing random genes with the same slightly-positive fitness distribution) is needed to distinguish true co-regulation from shared dispensability — see Interpretation section for full discussion.
By mechanism, efflux AMR genes show the strongest enrichment for amino acid biosynthesis (histidine in 6 organisms, tryptophan in 5), while metal resistance genes show stronger chemotaxis enrichment (4 organisms). However, no GO term is significantly mechanism-specific after FDR correction.
(Notebooks: 03b_enrichment_interproscan.ipynb; cf. 03_support_networks.ipynb for old-annotation null result)
3. Support networks are organism-specific, not mechanism-specific
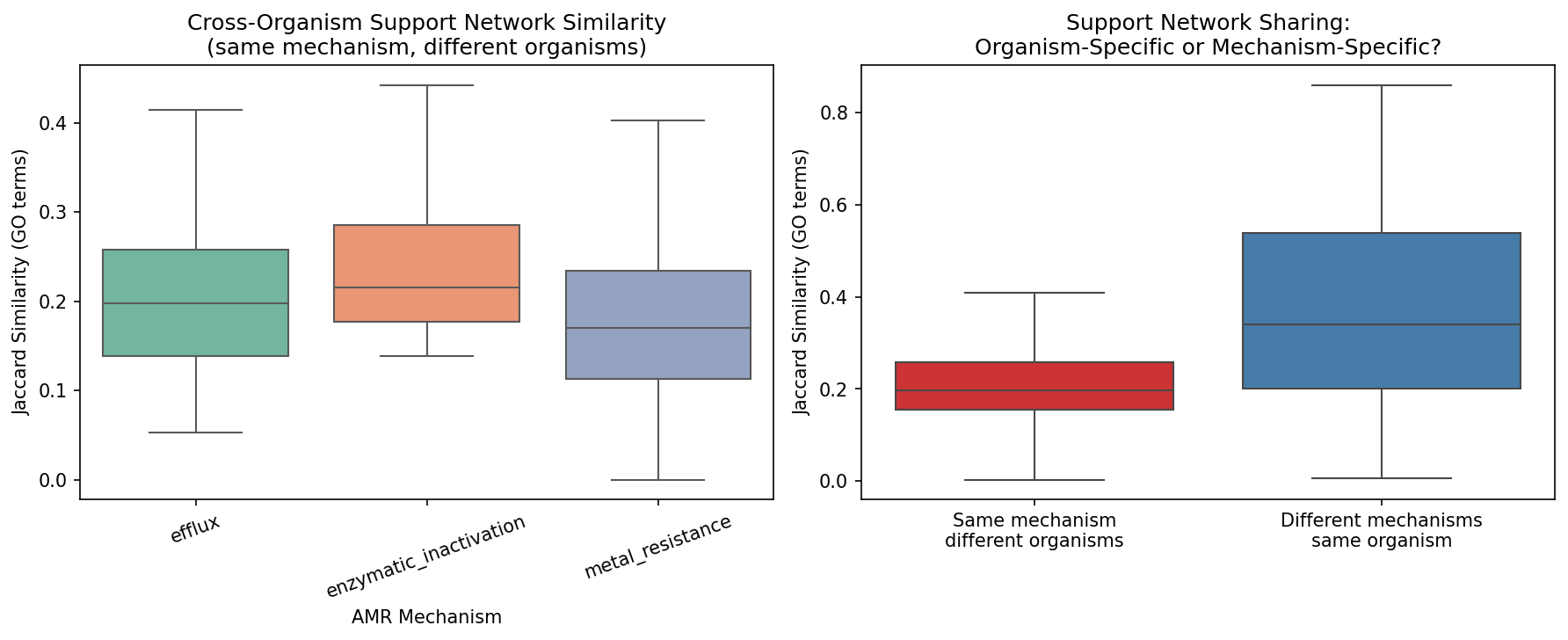
Different AMR mechanisms within the same organism share far more support partners than the same mechanism across organisms:
| Comparison | Mean Jaccard (GO) | Interpretation |
|---|---|---|
| Cross-mechanism (same organism) | 0.375 | Different AMR genes in the same bug share 38% of cofitness GO terms |
| Within-mechanism (across organisms) | 0.207 | Same mechanism in different bugs shares only 21% |
| MWU test | p = 4.3×10⁻¹³ | Highly significant difference |
This means the organism's regulatory landscape — its particular wiring of transcription, metabolism, and signaling — shapes the AMR support network far more than the type of resistance mechanism. An efflux pump in Pseudomonas shares more cofitness partners with a beta-lactamase in the same Pseudomonas than with an efflux pump in Shewanella.
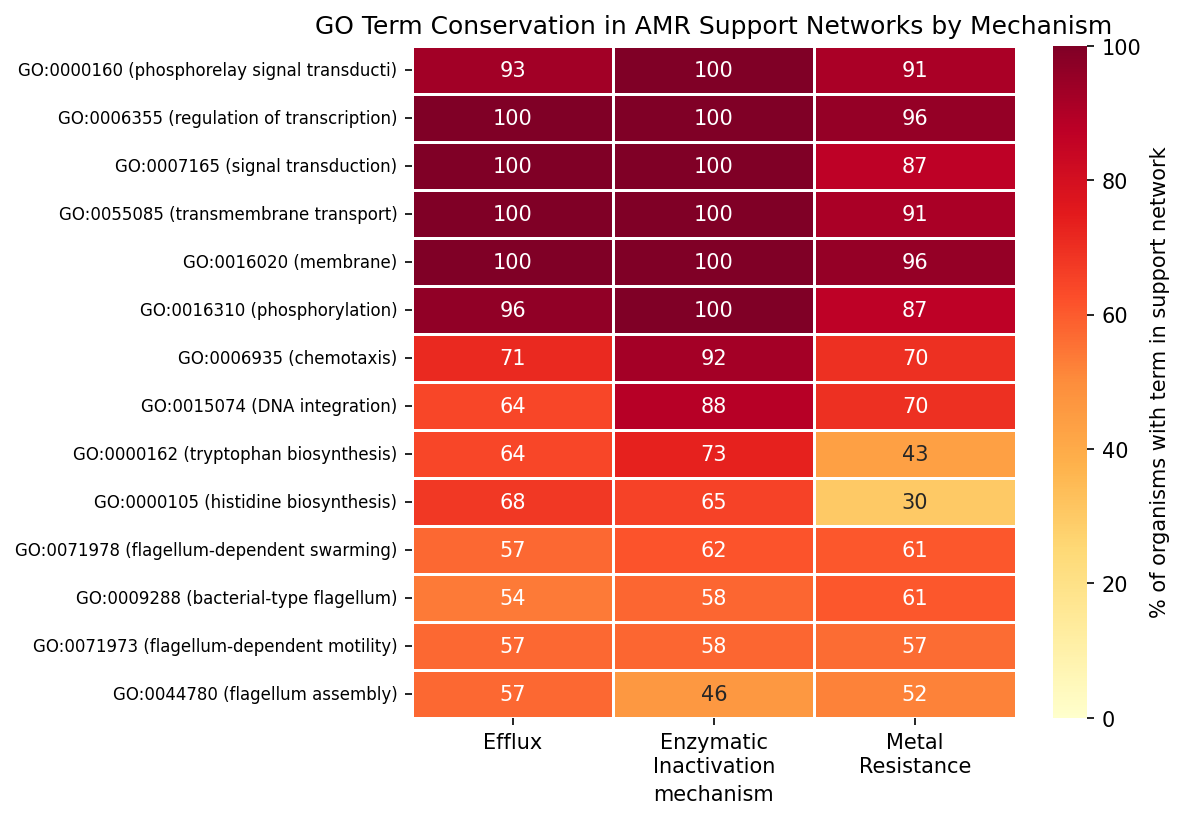
The conserved core across all mechanisms includes transmembrane transport (87–100% of organisms), signal transduction (87–100%), transcription regulation (96–100%), and phosphorelay signaling (91–100%). Flagellar motility (53–61%) and amino acid biosynthesis (30–73%) form a second tier of conservation. The only hint of mechanism specificity is histidine biosynthesis (efflux 68% vs metal 30%, p = 0.013 uncorrected, q = 0.18 after FDR).
(Notebooks: 04b_cross_organism_interproscan.ipynb; cf. 04_cross_organism_conservation.ipynb for old KEGG analysis)
4. Support network size does not predict fitness cost (H3 not supported)
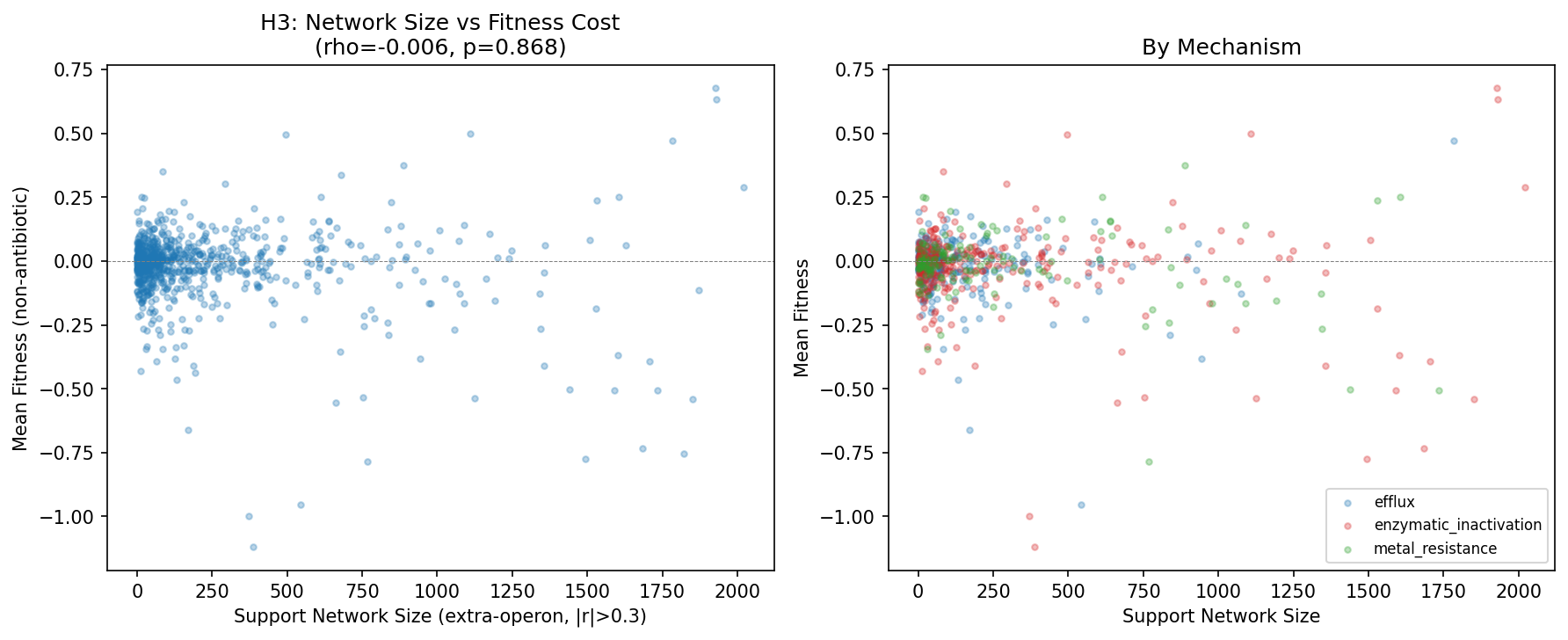
There is no correlation between cofitness support network size and AMR gene fitness cost (Spearman rho = −0.006, p = 0.87, N = 769). This holds within each mechanism (efflux rho = −0.049, enzymatic rho = +0.038, metal rho = −0.031; all p > 0.4). The uniform cost of resistance (+0.086 from amr_fitness_cost) is not explained by the size of the co-regulatory neighborhood.
(Notebook: 03_support_networks.ipynb)
5. Annotation quality is critical: InterProScan reveals what SEED/KEGG missed
The switch from old Fitness Browser SEED/KEGG annotations to InterProScan GO annotations on the same data transformed a null result into a significant finding:
| Analysis | Old Annotations (SEED/KEGG) | InterProScan GO |
|---|---|---|
| Coverage | 40–80% per organism, variable | 60% uniform across all genes |
| Enrichment tests significant (FDR<0.05) | 0 / 280 | 35 / 3,193 |
| GO terms in ≥3 organisms | 0 | 6 (flagella, amino acid biosynthesis) |
| Cross-organism Jaccard (within-mech) | 0.069 | 0.207 (3× higher) |
| Cross-organism Jaccard (cross-mech) | 0.249 | 0.375 (1.5× higher) |
This is a methodological contribution: genome-wide cofitness analyses require high-coverage, uniformly computed functional annotations to detect real biological signals. InterProScan on pangenome cluster representatives provides this.
(Notebooks: 03_support_networks.ipynb vs 03b_enrichment_interproscan.ipynb; 04_cross_organism_conservation.ipynb vs 04b_cross_organism_interproscan.ipynb)
Results
Data Assembly (NB01)
- 28 organisms with AMR genes, fitness matrices, and ICA modules (full intersection)
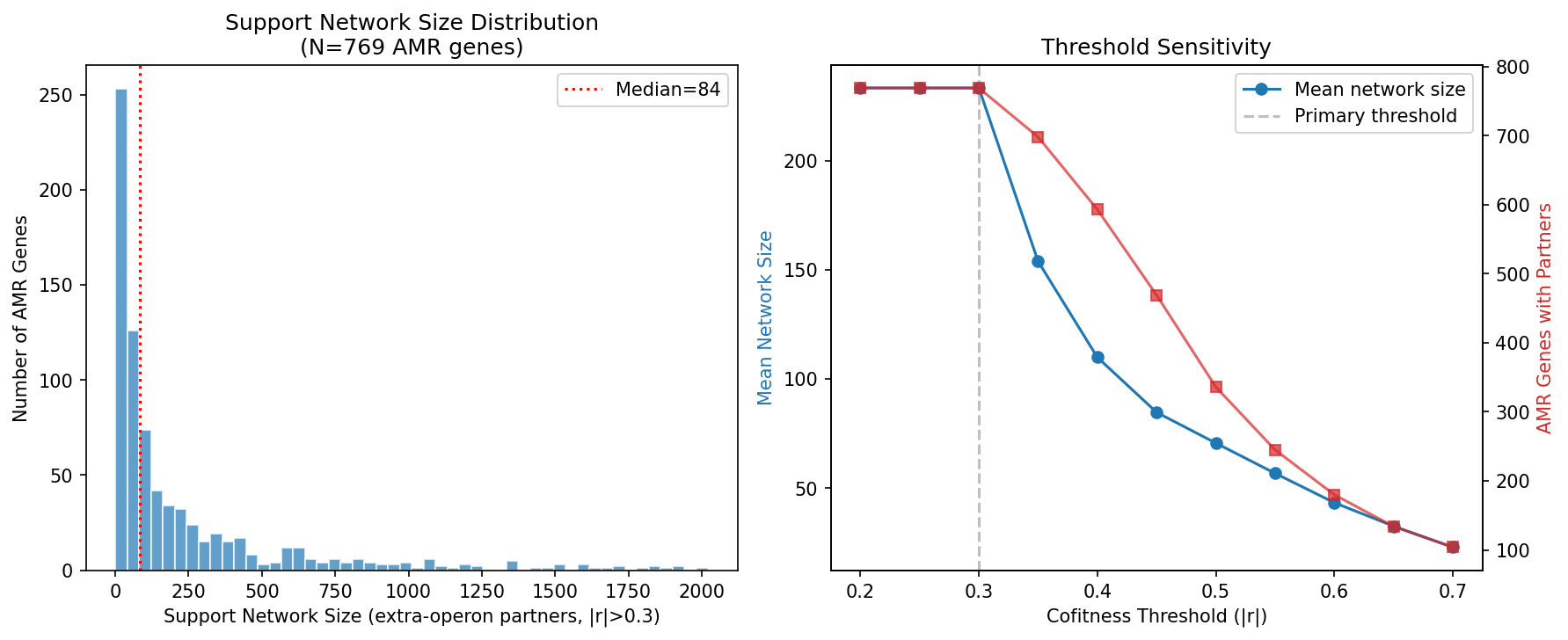
- 801 AMR genes with fitness data; 769 (96%) have ≥1 extra-operon cofitness partner at |r| > 0.3
- 180,370 total cofitness partners; 179,375 extra-operon (only 0.6% excluded as near-operon)
- Support network sizes: mean 233 genes (|r|>0.3), 110 (|r|>0.4), 71 (|r|>0.5)
Module Analysis (NB02)
- 24% of AMR genes in ICA modules (192/801)
- AMR-containing modules significantly larger (median 46 vs 27, p = 1.7×10⁻⁸)
- 136 unique module families contain AMR genes; 99% in cross-organism conserved families
Support Network Enrichment (NB03/NB03b)
| Source | Tests | Significant (FDR<0.05) | Top signal |
|---|---|---|---|
| Old SEED | 280 | 0 | None |
| InterProScan GO | 3,193 | 35 | Flagellar motility (5 orgs) |
| Permutation (old SEED) | 250 | 23 | Membrane/cell wall (24%, fold 1.12) |
| Mechanism-specific GO | 9,244 | 212 | Efflux: histidine biosynthesis (6 orgs) |
Cross-Organism Conservation (NB04/NB04b)
| Metric | Old KEGG | InterProScan GO |
|---|---|---|
| Within-mechanism Jaccard | 0.069 | 0.207 |
| Cross-mechanism Jaccard | 0.249 | 0.375 |
| Cross > Within p-value | 1.0 | 4.3×10⁻¹³ |
| Mechanism-specific terms (FDR<0.05) | 2 (lipoprotein, toluene tolerance) | 0 |
Interpretation
Cofitness enrichment may reflect shared dispensability, not co-regulation
The enrichment of flagellar motility, chemotaxis, and amino acid biosynthesis in AMR support networks admits two interpretations:
Interpretation A (co-regulation): AMR genes are genuinely embedded in regulatory programs that co-control resistance, motility, and biosynthesis, and the cofitness signal reflects condition-specific co-regulation via shared transcription factors or signaling cascades.
Interpretation B (shared dispensability): The enrichment is an artifact of the Fitness Browser experimental design. Nearly all FB experiments use shaken liquid culture, where flagella are useless (no surface to swim on, no chemical gradient to follow). Most use rich or defined media with amino acid supplements, where biosynthesis is redundant. AMR genes are similarly dispensable without antibiotics. All three gene classes — AMR, flagellar, and biosynthetic — are therefore metabolic burdens under standard lab conditions, producing modestly positive knockout fitness across most experiments. The cofitness signal reflects shared membership in the "dispensable under lab conditions" gene class, not a mechanistic connection.
Evidence favoring Interpretation B:
- The enriched categories are precisely the ones that would be dispensable in shaken flask culture: motility (useless without surfaces), chemotaxis (useless without gradients), amino acid biosynthesis (redundant in supplemented media)
- Energy metabolism is NOT enriched (0/25 organisms, fold 0.91 from the permutation test) — because energy metabolism IS useful in lab conditions
- The permutation test matched random genes by conservation class but not by mean fitness level. If we had drawn random genes with the same slightly-positive fitness distribution as AMR genes, the flagellar enrichment might disappear entirely
What Pearson correlation does and doesn't control: Pearson r subtracts each gene's mean fitness before correlating, so two genes that are uniformly slightly positive would show zero correlation. The cofitness signal requires condition-specific covariation. However, dispensable genes may share condition-responsive patterns (e.g., both are more dispensable under nutrient-rich conditions and less dispensable under starvation) without being co-regulated — they just respond similarly to the same environmental axis.
The key unresolved test: A fitness-matched permutation — drawing random non-AMR genes with the same mean fitness distribution (−0.05 to +0.05) — would distinguish the two interpretations. If random slightly-positive genes also show flagellar enrichment in their cofitness neighborhoods, Interpretation B is correct. If AMR genes uniquely enrich for flagella even among genes with similar fitness, Interpretation A is supported. This is the single most important follow-up analysis.
Organism regulatory architecture trumps resistance mechanism (robust finding)
The organism-specificity finding is robust to the dispensability confound: regardless of why AMR genes correlate with their partners, the fact that different AMR mechanisms within the same organism share more partners (J = 0.375) than the same mechanism across organisms (J = 0.207, p = 4.3×10⁻¹³) reveals that each organism has its own characteristic set of dispensable/co-varying genes. This is a genuine structural finding about how genomes are organized, not an artifact of the enrichment categories.
This explains a puzzle from amr_fitness_cost: mechanism predicts conservation (metal 44% accessory vs efflux 13%) but not cost. The cost is determined by the organism's gene content and growth context, while conservation is determined by the mechanism's acquisition history.
AMR genes are in larger-than-average modules (robust finding)
The ICA module size result (AMR modules median 46 vs non-AMR 27, p = 1.7×10⁻⁸) is also robust — ICA decomposition captures condition-specific co-regulation, not just shared mean fitness. AMR genes that do land in modules are in larger, cross-organism conserved modules, indicating they are embedded in broad cellular programs with genuine co-regulatory structure.
Literature Context
- Sagawa et al. (2017) validated that cofitness recovers transcriptional regulatory relationships in 24 bacteria. Our cofitness analysis successfully identifies functional categories — the question is whether the AMR-motility signal reflects regulation or shared dispensability.
- Martinez & Rojo (2011) reviewed the linkage between metabolism and intrinsic resistance, noting that global metabolic regulators modulate antibiotic susceptibility. If Interpretation A is correct, this framework directly supports our findings.
- Olivares Pacheco et al. (2017) showed efflux pump costs in P. aeruginosa are metabolic (PMF drain) and compensated by metabolic rewiring. The PMF competition model would explain why efflux genes correlate with flagellar genes (both use PMF) — but shared dispensability could produce the same signal.
- Eckartt et al. (2024, Nature) identified compensatory mutations targeting the same functional pathway as the resistance mutation. Our organism-specificity finding is consistent with this: compensation integrates AMR genes into whatever programs the organism already has.
- Nichols et al. (2011, Cell) showed that condition-dependent fitness profiles reveal gene function in E. coli. The general principle holds, but our analysis highlights a caveat: functional enrichment in cofitness neighborhoods can reflect shared experimental context (lab conditions) rather than biological co-regulation.
Novel Contribution
This is the first pan-bacterial mapping of AMR co-fitness neighborhoods across 28 organisms. Key contributions:
1. AMR cofitness neighborhoods are enriched for flagellar motility and amino acid biosynthesis — but this may reflect shared dispensability under lab conditions rather than mechanistic co-regulation (a key caveat for all cofitness-based functional inference)
2. Support networks are organism-specific (J = 0.375) not mechanism-specific (J = 0.207) — a robust structural finding about genome organization
3. Annotation quality is critical: InterProScan GO transformed a null result into a significant one, demonstrating that legacy annotations are insufficient for functional genomics
4. AMR genes are in larger-than-average ICA modules (p = 1.7×10⁻⁸), confirming they are embedded in broad cellular programs
5. The analysis identifies a general caveat for cofitness-based functional genomics: genes that are dispensable under experimental conditions may show artifactual co-regulation, a concern relevant beyond AMR to any cofitness study using lab-grown organisms
Limitations
- Shared dispensability confound (critical): The flagellar/biosynthesis enrichment may reflect shared "useless under lab conditions" status rather than mechanistic co-regulation. AMR genes (no antibiotics present), flagellar genes (shaken liquid culture), and amino acid biosynthesis genes (supplemented media) are all metabolic burdens under FB experimental conditions. A fitness-matched permutation test (matching on mean fitness level, not just conservation) is needed to resolve this.
- Cofitness ≠ co-regulation: Even without the dispensability confound, high cofitness implies shared fitness phenotypes, not direct transcriptional co-regulation. Additionally, the cofitness computation treats missing fitness values as zero in z-score space (via
np.nan_to_num), which approximates but does not equal pairwise-complete Pearson correlation. For the relatively dense FB matrices this is unlikely to affect conclusions. - GO term granularity: Broad GO terms (transmembrane transport, membrane) appear in nearly all support networks and all genomes, potentially masking more specific signals.
- Support network size: At |r| > 0.3, mean network size is 233 genes (large). This includes many weak associations. Confirmation at |r| > 0.4 is needed.
- H3 null result: The absence of a network-size-cost correlation may reflect insufficient variance in fitness cost across genes.
- FB organism bias: The 28 organisms are lab-adapted, phylogenetically biased (many Pseudomonas), and limited in ecological diversity.
- Operon exclusion heuristic: The 5-ORF exclusion zone uses matrix row index position as a proxy for genomic proximity, which may not reliably reflect chromosomal gene order. Genomic coordinate columns (
begin,end,strand) are loaded in the annotation data and could be used for a proper coordinate-based exclusion in future work. Only 0.6% of pairs are excluded by the current heuristic.
Future Directions
- Fitness-matched permutation (critical): The most important follow-up is a permutation test drawing random non-AMR genes matched on mean fitness level (not just conservation class). If random genes with fitness between −0.05 and +0.05 also show flagellar/biosynthesis enrichment in their cofitness neighborhoods, the enrichment is a shared-dispensability artifact. If AMR genes uniquely enrich even among fitness-matched genes, the co-regulation interpretation is supported.
- Condition-specific cofitness: Computing cofitness separately for antibiotic conditions vs standard growth could distinguish true co-regulation from shared dispensability. If AMR-flagella cofitness is specifically elevated under antibiotic stress (not just standard growth), it would support a regulatory connection.
- Verify flagellar gene dispensability directly: Check whether flagellar gene knockouts show positive mean fitness across FB experiments. If so, the shared-dispensability explanation is confirmed as plausible. If flagellar knockouts are near-zero or negative, the co-regulation interpretation is strengthened.
- Pfam domain-level enrichment: Pfam has 88% coverage (vs 68% for GO). Domain-level enrichment may reveal more specific support network functions than broad GO terms.
- Extend to other dispensable gene classes: Test whether phage defense genes, secondary metabolite genes, or other "condition-specific" gene classes show the same cofitness patterns as AMR genes. If so, the signal is a general feature of dispensable genes under lab conditions, not AMR-specific.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
kbase_ke_pangenome |
interproscan_go, interproscan_domains (Pfam), bakta_annotations |
GO terms, Pfam domains, KEGG orthology for FB gene clusters |
kescience_fitnessbrowser |
(via cached matrices) | Fitness profiles for cofitness computation |
| Cross-project | amr_fitness_cost/data/amr_genes_fb.csv |
AMR gene catalog |
| Cross-project | amr_fitness_cost/data/amr_fitness_noabx.csv |
Per-gene fitness costs |
| Cross-project | fitness_modules/data/modules/ |
ICA module membership |
| Cross-project | fitness_modules/data/module_families/ |
Cross-organism module families |
| Cross-project | fitness_modules/data/matrices/ |
Cached fitness matrices |
| Cross-project | conservation_vs_fitness/data/fb_pangenome_link.tsv |
FB → pangenome cluster mapping |
Generated Data
| File | Rows | Description |
|---|---|---|
data/amr_cofitness_partners.csv |
180,370 | All cofitness partners ( |
data/amr_module_membership.csv |
818 | AMR gene → ICA module assignments |
data/amr_modules_characterized.csv |
209 | AMR-containing modules with properties |
data/fb_interproscan_go.csv |
438,000 | InterProScan GO terms for FB gene clusters |
data/fb_interproscan_pfam.csv |
228,672 | InterProScan Pfam domains for FB gene clusters |
data/fb_bakta_kegg.csv |
41,611 | Bakta KEGG orthology for FB gene clusters |
data/go_enrichment_interproscan.csv |
3,193 | Per-organism GO enrichment results |
data/mechanism_go_enrichment.csv |
9,244 | Per-mechanism GO enrichment results |
data/go_conservation_by_mechanism.csv |
1,078 | GO term presence by mechanism × organism |
data/hub_support_genes.csv |
47,327 | Hub genes (partner of multiple AMR genes) |
data/jaccard_go_within_mechanism.csv |
956 | Cross-organism Jaccard (GO terms, within-mechanism) |
data/jaccard_go_cross_mechanism.csv |
71 | Within-organism Jaccard (GO terms, cross-mechanism) |
References
- Cox G, Wright GD. (2013). "Intrinsic antibiotic resistance: mechanisms, origins, challenges and solutions." Int J Med Microbiol 303(6-7):287-292. PMID: 23499305
- Eckartt KA, et al. (2024). "Compensatory evolution in NusG improves fitness of drug-resistant M. tuberculosis." Nature 627:186-194. PMID: 38509362
- Martinez JL, Rojo F. (2011). "Metabolic regulation of antibiotic resistance." FEMS Microbiol Rev 35(5):768-789. PMID: 21645016
- Olivares Pacheco J, et al. (2017). "Metabolic compensation of fitness costs is a general outcome for antibiotic-resistant Pseudomonas aeruginosa mutants overexpressing efflux pumps." mBio 8(4):e00500-17. PMID: 28765215
- Patel A, Matange N. (2021). "Adaptation and compensation in a bacterial gene regulatory network evolving under antibiotic selection." eLife 10:e70931. PMID: 34591012
- Price MN, et al. (2018). "Mutant phenotypes for thousands of bacterial genes of unknown function." Nature 557(7706):503-509. PMID: 29769716
- Sagawa S, et al. (2017). "Validating regulatory predictions from diverse bacteria with mutant fitness data." PLoS ONE 12(5):e0178258. PMID: 28542589
- Arkin AP, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nature Biotechnology 36(7):566-569. PMID: 29979655
Discoveries
Cofitness analysis of 801 AMR genes across 28 organisms reveals that AMR cofitness neighborhoods are enriched for flagellar motility (GO:0071973, 5 organisms FDR<0.05), flagellum assembly (GO:0044780, 5 orgs), histidine biosynthesis (GO:0000105, 3 orgs), and tryptophan biosynthesis (GO:0000162, 3 or
Read more →Data Collections
Review
Summary
This is a scientifically rigorous and well-executed follow-up to amr_fitness_cost, mapping the co-fitness neighborhoods of 801 AMR genes across 28 bacteria using full pairwise Pearson correlations from cached fitness matrices and ICA fitness modules. The project stands out for its scientific honesty: the REPORT provides an unusually thorough self-critique, identifying the "shared dispensability" confound as a plausible alternative explanation for the central finding (flagellar/biosynthetic enrichment) and proposing the exact experiment needed to resolve it. The four pre-specified hypotheses (H1–H4) are tested systematically, and negative results (H2 partially unsupported, H3 not supported) are presented alongside positive ones. The annotation quality comparison — where InterProScan GO transforms a null result (0/280 significant with old SEED) into a significant one (35/3,193 significant) — is a concrete methodological contribution. All 6 notebooks are executed with saved outputs, 9 figures cover every analysis stage, and 18 data files are present and internally consistent. The main gaps are (1) the InterProScan annotation extraction code exists only as commented-out Spark SQL in NB03b Cell 2, not as an executable cell; (2) the operon exclusion heuristic uses matrix index position rather than actual genomic coordinates; and (3) the permutation test matches on conservation class but not mean fitness level — a limitation the authors themselves identify as "the single most important follow-up analysis."
Methodology
Research question and hypotheses: The RESEARCH_PLAN pre-specifies four hypotheses (H1–H4) with explicit statistical tests (Fisher's exact with BH-FDR, Mann-Whitney U, Spearman correlation, permutation test), effect directions, expected outcomes, and seven named confounders. This level of pre-registration is well above average for exploratory bioinformatics projects and makes the analysis much easier to evaluate.
Literature context: The RESEARCH_PLAN cites 7 relevant papers with PMIDs, grounding the analysis in established cofitness validation (Sagawa et al. 2017), compensatory evolution theory (Eckartt et al. 2024, Patel & Matange 2021), and the intrinsic resistome concept (Cox & Wright 2013). The gap statement — that no study has systematically mapped AMR co-fitness neighborhoods pan-bacterially — is well-defined and appropriately scoped.
Approach: The decision to compute full pairwise cofitness from cached fitness matrices rather than using the FB cofit table (which stores only top ~96 partners) is correctly motivated and necessary for threshold-based network definition. The 28-organism intersection is verified with explicit locusId overlap checks showing 100% match in NB01 Cell 3. The three-threshold sensitivity analysis (|r| > 0.3, 0.4, 0.5) is good experimental design.
Data provenance: Six upstream tables and three prior projects are clearly identified. The cross-project dependency chain (amr_fitness_cost → fitness_modules → conservation_vs_fitness) is documented in the README with specific file paths.
Reproducibility: The README includes a clear 6-step reproduction guide with estimated runtimes per notebook, prerequisites, and a note about which notebook requires Spark. The requirements.txt file is present with version lower bounds. However, the InterProScan annotation files (fb_interproscan_go.csv, fb_interproscan_pfam.csv, fb_bakta_kegg.csv) are loaded by NB03b and NB04b but produced by Spark queries that appear only as commented-out code in NB03b Cell 2. While the comments do contain the full SQL queries, they are not executable as-is — a new user would need to uncomment and adapt them manually.
Code Quality
Notebook organization: All six notebooks follow a consistent structure: markdown header → imports/paths → data loading → computation → visualization → summary/save. Markdown section headers break each notebook into logical stages. The code is clean, uses descriptive variable names, and includes inline print statements that document intermediate results.
Pitfall awareness (checked against docs/pitfalls.md):
- locusId type mismatch: Handled with .astype(str) throughout — NB01 Cell 3 converts both sides before overlap checks, NB01 Cell 8 does the same for matrix indices. This is the single most commonly documented pitfall and it is handled correctly everywhere.
- FB numeric string columns: pd.to_numeric(errors='coerce') applied to fitness matrices in NB01 Cell 8.
- cofit table limitation: Correctly identified in both RESEARCH_PLAN and NB01 markdown. The full pairwise computation is the right approach.
- Row-wise apply performance: NB03b Cell 4 uses .apply(lambda r: ..., axis=1) on 179K rows to map (orgId, partner_locusId) tuples to gene_cluster_id. Per docs/pitfalls.md, this is orders of magnitude slower than a merge-based approach. Not a correctness issue, but affects runtime.
Cofitness computation (NB01 Cell 8): The numpy-vectorized approach is efficient and correct in structure: center each gene's profile, normalize, dot-product. The np.nan_to_num(..., 0) step replaces missing experiment values with zero in z-score space before computing correlations. This approximates but does not equal pairwise-complete Pearson correlation — missing data contributes zero to the numerator while the denominator reflects the full experiment count. The REPORT Limitations section (item 2) acknowledges this approximation and notes it is unlikely to affect conclusions for the relatively dense FB matrices.
Operon exclusion (NB01 Cell 8): The 5-ORF exclusion uses abs(locus_to_idx[partner] - locus_to_idx[amr_locus]) <= 5 — matrix index position as a proxy for genomic proximity. This is approximate: the matrix row order reflects how genes were loaded from the fitness matrix CSV, which is not guaranteed to match chromosomal gene order. Only 995/180,370 pairs (0.6%) are excluded, which is suspiciously low for 28 genomes and could indicate the heuristic is not reliably identifying proximal genes. The genomic coordinates (begin, end, scaffoldId, strand) are already loaded in NB01 Cell 12 but are not used. Both the RESEARCH_PLAN and REPORT acknowledge this approximation.
Module coverage label (NB01 Cell 3): The pct_in_module_file column and the final print statement ("In module file index: 801/801") check whether AMR locusIds appear in the module membership file index (which contains ALL genes, not just those assigned to modules). The variable is correctly labeled pct_in_module_file with a clarifying comment, but the final print line says "Module coverage: 100.0%" which could mislead. The correct 24% figure is computed independently in NB01 Cell 5 and NB02.
Permutation test (NB03 Cell 8): Runs 200 permutations instead of the planned 1,000. A code comment explains the reduction. For the main conclusion (no significant SEED enrichment), this is adequate. The null result at 200 draws would remain null at 1,000.
Sensitivity analysis at |r| > 0.4 (NB03b Cell 10): This is a strength. The notebook runs the full GO enrichment at the stricter |r| > 0.4 threshold and finds the signal is actually stronger: flagellum assembly significant in 8 organisms (vs 5 at |r| > 0.3), flagellum-dependent motility in 7 organisms (vs 5), tryptophan biosynthesis in 6 organisms (vs 3). This confirms the enrichment is not driven by a tail of weak correlations and significantly strengthens the main finding.
Statistical methods: Fisher's exact test with BH-FDR correction is appropriate for the enrichment analyses. The FDR scope (within each organism for NB03/NB03b, within each mechanism-organism pair for mechanism-specific tests) is correctly applied and matches the RESEARCH_PLAN specification. Mann-Whitney U for module size comparisons and Spearman for the network-size-vs-cost correlation are both appropriate non-parametric choices.
Findings Assessment
H1 (AMR support networks enriched for specific functions): Supported with InterProScan GO. Six GO terms significant in ≥3 organisms (FDR < 0.05): four flagellar terms (motility, assembly, swarming, bacterial-type flagellum; 4–5 organisms, mean OR 4.7–5.3) and two amino acid biosynthesis terms (histidine, tryptophan; 3 organisms, OR 5.3). The comparison with old SEED annotations (0/280 significant) is properly retained as a baseline. The sensitivity check at |r| > 0.4 confirms and strengthens these results. Crucially, the REPORT provides an intellectually honest discussion of the shared dispensability confound: flagella are useless in shaken liquid culture, biosynthesis is redundant in supplemented media, and AMR genes are dispensable without antibiotics — all three gene classes are metabolic burdens under FB experimental conditions. The proposed follow-up (fitness-matched permutation) is exactly the right test.
H2 (Efflux in stress modules, enzymatic in isolated modules): Partially tested. AMR-containing modules are larger than non-AMR modules (median 46 vs 27, p = 1.7×10⁻⁸) and 99% are in cross-organism conserved families. But efflux vs enzymatic module sizes are indistinguishable (both median 48, MWU p = 0.91). H2 as formulated is not supported. The REPORT correctly frames this as a partial result.
H3 (Network size predicts fitness cost): Not supported (Spearman rho = −0.006, p = 0.87, N = 769). Holds within each mechanism. Properly contextualized as possibly reflecting narrow cost variance (std = 0.15) rather than absence of a real relationship.
H4 (Support networks conserved across organisms): The organism-specificity finding is the project's most robust result. Cross-mechanism Jaccard (same organism) = 0.375 vs within-mechanism Jaccard (different organisms) = 0.207, MWU p = 4.3×10⁻¹³. As the REPORT notes, this finding is robust to the dispensability confound because it is a structural observation about genome co-regulation architecture. The finding also connects back to amr_fitness_cost: mechanism predicts conservation but not cost, and now we see that the cost context is organism-determined.
Annotation quality lesson: The SEED → InterProScan comparison (0/280 vs 35/3,193 significant enrichments; Jaccard 0.069 vs 0.207 for cross-organism conservation) is a genuine methodological contribution. The 68% GO coverage vs variable SEED coverage explains why prior analyses may have missed functional signals in cofitness data.
Limitations: Seven specific limitations are documented in the REPORT, prioritized appropriately. The shared dispensability discussion is genuinely insightful and has implications beyond AMR — it identifies a general caveat for all cofitness-based functional inference using lab-grown organisms.
Completeness: No incomplete analyses or placeholder text. All 18 data files listed in REPORT.md are present in data/. All 9 figures referenced in the REPORT are present in figures/. The three-document structure (README → RESEARCH_PLAN → REPORT) is well-organized and internally consistent.
Suggestions
-
[Critical — Reproducibility] Make the InterProScan data extraction executable. NB03b Cell 2 contains the Spark SQL queries as comments but they cannot be run as-is. Convert to an executable cell guarded by
if not os.path.exists(os.path.join(DATA, 'fb_interproscan_go.csv')):so the notebook either regenerates annotations from Spark (on JupyterHub) or loads cached files (locally). This is the single most important reproducibility gap — without it, a new user cannot reproduce NB03b or NB04b from scratch. -
[Moderate — Methodology] Replace the index-position operon exclusion with a coordinate-based one. The
begin,end,scaffoldId, andstrandcolumns are already loaded in NB01 Cell 12 — build a per-organism lookup of{locusId: (scaffoldId, begin, end, strand)}and exclude partners within a defined chromosomal distance (e.g., 10 kb on the same scaffold and strand). The current heuristic excludes only 0.6% of pairs, which may reflect that index order does not match chromosomal order. A proper coordinate-based exclusion would give more confidence that the support networks are not confounded by polar effects. -
[Moderate — Methodology] Run the fitness-matched permutation test identified as "the single most important follow-up" in the REPORT. Drawing random non-AMR genes matched on mean fitness level (−0.05 to +0.05) and comparing their cofitness neighborhoods' GO enrichment to AMR genes would directly resolve the shared-dispensability question. This could be added as a cell in NB03b without a new notebook.
-
[Minor — Code] Fix the misleading "Module coverage: 100.0%" print statement in NB01 Cell 3. The variable is already named
pct_in_module_filewith a clarifying comment, but the final print line still says "Module coverage." Change to:print(f'In module file index: {org_stats_df["amr_in_modules"].sum()}/{org_stats_df["amr_in_matrix"].sum()} (note: file index coverage, not module assignment)'). -
[Minor — Performance] Replace
.apply(lambda r: ..., axis=1)in NB03b Cell 4 with a merge-based lookup perdocs/pitfalls.md. Create a DataFrame from thelocus_to_clusterdict and merge on['orgId', 'partner_locusId']— this would be orders of magnitude faster on 179K rows. -
[Minor — Methods precision] Add a brief note in the REPORT Methods or a code comment in NB01 Cell 8 explicitly stating that missing experiment values are replaced with zero in z-score space (
np.nan_to_num) and that this approximates pairwise-complete Pearson correlation. The current Limitations item 2 mentions this but it could be more prominent. -
[Nice-to-have — Analysis] Leverage the already-loaded Pfam annotations (
fb_interproscan_pfam.csv, 88% coverage, 229K rows) for domain-level enrichment analysis. Pfam's higher coverage and more specific domain-level vocabulary could reveal support network functions masked by broad GO terms like "membrane" and "transmembrane transport." The REPORT Future Directions item 4 identifies this as a next step, and the data is already in hand. -
[Nice-to-have — Verification] Directly check whether flagellar gene knockouts show positive mean fitness across FB experiments, as proposed in REPORT Future Directions item 3. This would take ~10 lines of code in NB03b: load all gene fitness values, filter to genes annotated with GO:0071973 (flagellar motility), and plot their mean fitness distribution. If the distribution overlaps with AMR gene fitness, the shared-dispensability interpretation is strengthened.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Amr Module Analysis
Amr Module Coverage
Cofitness Threshold Sensitivity
Conserved Kegg By Mechanism
Go Conservation Heatmap
Go Enrichment Interproscan
Jaccard Go Comparison
Network Size Vs Fitness
Support Network Enrichment
Notebooks
01_data_assembly.ipynb
01 Data Assembly
View notebook →
02_amr_in_modules.ipynb
02 Amr In Modules
View notebook →
03_support_networks.ipynb
03 Support Networks
View notebook →
03b_enrichment_interproscan.ipynb
03B Enrichment Interproscan
View notebook →
04_cross_organism_conservation.ipynb
04 Cross Organism Conservation
View notebook →
04b_cross_organism_interproscan.ipynb
04B Cross Organism Interproscan
View notebook →