Acinetobacter baylyi ADP1 Data Explorer
CompletedResearch Question
What is the scope and structure of a comprehensive ADP1 database, and how do its annotations, metabolic models, and phenotype data intersect with BERDL collections (pangenome, biochemistry, fitness, PhageFoundry)?
Research Plan
Hypothesis
This is an exploration project rather than a hypothesis-driven study. The working assumption is that the ADP1 database contains identifiers (reaction IDs, pangenome cluster IDs, UniRef IDs, COG/KEGG annotations) that map directly to BERDL collections, enabling cross-referencing between user-provided experimental data and the broader lakehouse.
Approach
Phase 1: Database Inventory
Characterize every table in berdl_tables.db:
- Row counts, column types, NULL coverage
- Key identifier formats and value distributions
- Relationships between tables (shared keys)
Phase 2: BERDL Connection Mapping
For each identifier type found in the ADP1 database, check whether it maps to a BERDL collection:
| ADP1 Identifier | BERDL Collection | Link Strategy |
|---|---|---|
reaction_id (rxn00001 format) |
kbase_msd_biochemistry.reaction |
Direct match on reaction ID |
pangenome_cluster_id |
kbase_ke_pangenome.gene_cluster |
Match via species clade + cluster ID |
uniref_50/90/100 |
kbase_uniref50/90/100 |
Direct match on UniRef ID |
cog |
kbase_ke_pangenome.eggnog_mapper_annotations |
Match via COG_category |
ko |
kbase_ke_pangenome.eggnog_mapper_annotations |
Match via KEGG_ko |
genome_id (GCF/GCA format) |
kbase_ke_pangenome.genome |
Direct match on genome_id |
| Organism name | kescience_fitnessbrowser.organism |
Taxonomy match |
| Organism name | phagefoundry_acinetobacter_genome_browser |
Strain match |
Phase 3: Integration Highlights
For the strongest connection points, pull sample data from both sides and demonstrate the join works.
Revision History
- v1 (2026-02-18): Initial plan
Overview
This project explores a user-provided SQLite database (berdl_tables.db, 136 MB) containing comprehensive data for Acinetobacter baylyi ADP1 and related genomes. The database includes 15 tables spanning genome features (5,852 genes with 51 annotation columns), metabolic model reactions (17,984), gene-phenotype associations (239K), essentiality classifications, proteomics, mutant growth data, and a 14-genome pangenome (43K features).
The goal is to (1) characterize what's in the database, (2) identify connection points to BERDL collections, and (3) demonstrate the user_data/ convention for bringing external data into observatory projects.
Key Findings
1. Rich Multi-Omics Database with 6 Data Modalities
The user-provided SQLite database contains 15 tables with 461,522 total rows and 135 MB of data for Acinetobacter baylyi ADP1 and 13 related genomes. The central genome_features table has 5,852 genes with 51 annotation columns spanning six data modalities: TnSeq essentiality (58% coverage), FBA metabolic flux (15%), mutant growth fitness on 8 carbon sources (39%), proteomics across 7 strains (41%), pangenome classification (54%), and functional annotations via COG/KO/Pfam/UniRef (34-55%).
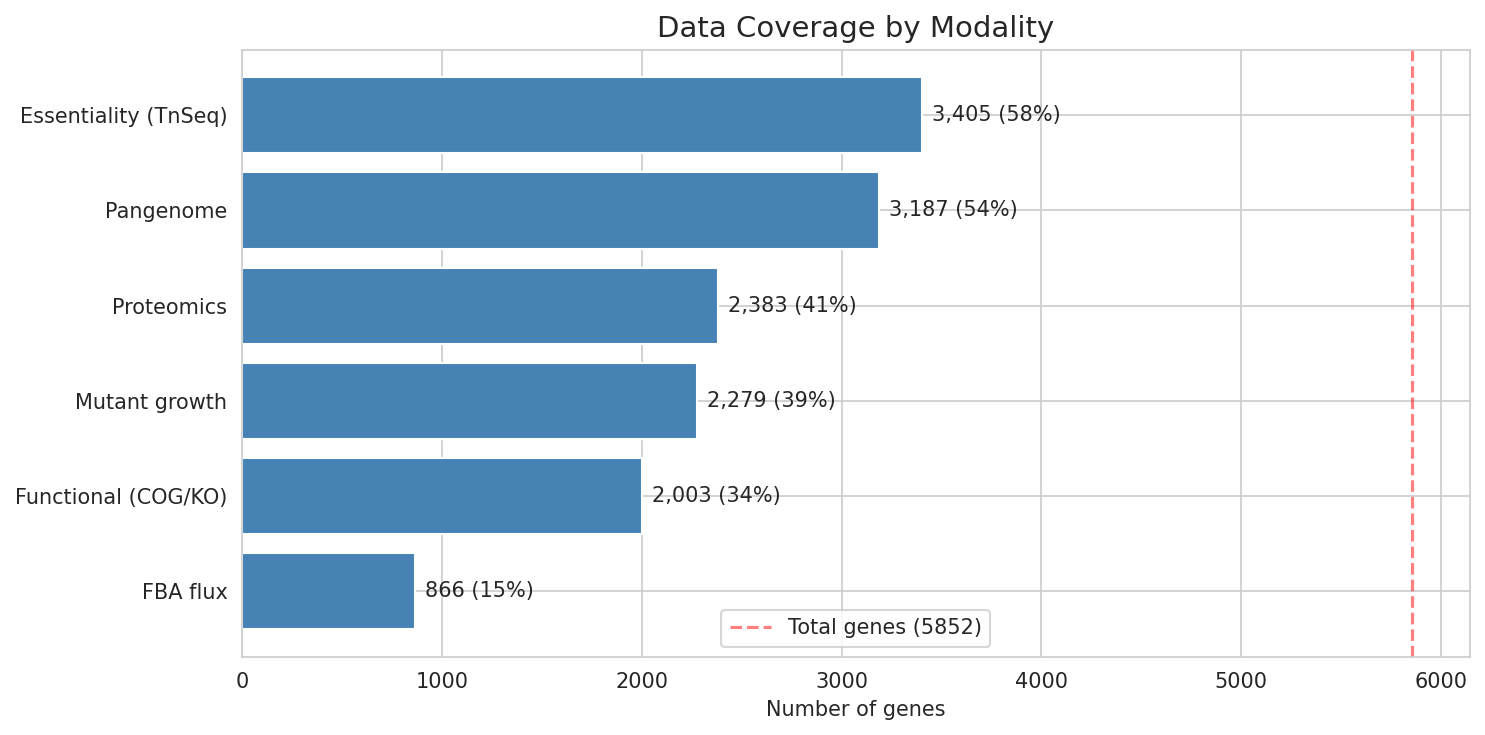
No single gene has data across all six modalities simultaneously, but the pairwise overlaps are substantial — particularly between essentiality, pangenome, and proteomics.
(Notebook: 01_database_exploration.ipynb)
2. Strong BERDL Connectivity: 4 of 5 Connection Types at >90% Match
Querying BERDL via Spark validated that the ADP1 database connects strongly to BERDL collections:
| Connection | ADP1 Count | BERDL Matches | Match % |
|---|---|---|---|
| Genome IDs → Pangenome | 13 | 13 | 100% |
| Reactions → Biochemistry | 1,330 | 1,210 | 91% |
| Compounds → Biochemistry | 230 | 230 | 100% |
| Cluster IDs → Pangenome | 4,891 | 4,891 | 100% (via mapping) |
| ADP1 → Fitness Browser | 1 | 0 | Not present |
All 13 BERDL-format genomes belong to the species s__Acinetobacter_baylyi (clade s__Acinetobacter_baylyi--RS_GCF_000368685.1), which has a BERDL pangenome of 3,207 core and 1,684 accessory gene clusters. The 120 unmatched reactions (9%) may represent custom or draft reactions not yet in ModelSEED. ADP1 is absent from the Fitness Browser, making the mutant growth data in this database a unique resource not available elsewhere in BERDL.
(Notebook: 02_berdl_connection_scan.ipynb)
3. Pangenome Cluster ID Bridge: 100% Mapping via Gene Junction Table
The ADP1 database uses mmseqs2-style cluster IDs (NHSXFYEX_mmseqsCluster_NNNN) while BERDL uses centroid gene IDs (NC_005966.1_1024) — completely different naming conventions with 0% direct string match. The bridge was found through BERDL's gene_genecluster_junction table: BERDL cluster IDs map to member gene IDs, which match the feature_id column in the ADP1 pan_genome_features table, which in turn carries the ADP1-style cluster_id.
All 4,891 BERDL clusters mapped successfully to 4,081 unique ADP1 clusters (100% gene-level match across 43,754 genes). The saved mapping table (data/cluster_id_mapping.csv) enables joining any BERDL pangenome annotation (eggNOG, functional predictions) to ADP1 genes.
(Notebook: 03_cluster_id_mapping.ipynb)
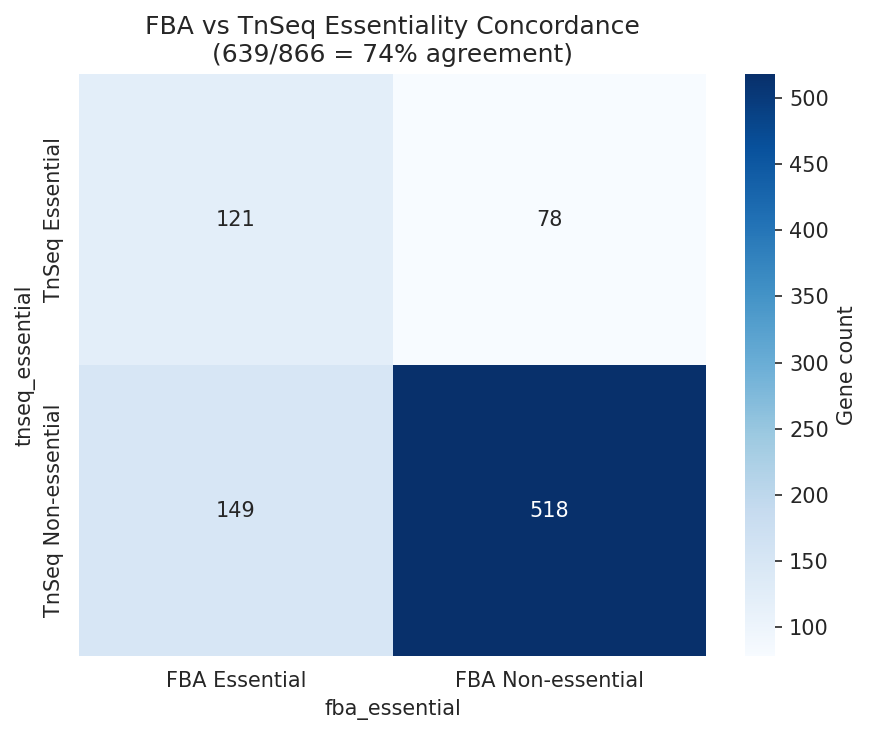
4. FBA and TnSeq Essentiality Agree 74% of the Time
Of 866 genes with both FBA flux predictions and TnSeq essentiality calls, 639 (73.8%) are concordant. The discordant 26% — genes where the metabolic model and experimental data disagree — are candidates for model refinement or may reflect regulatory effects not captured by FBA.
Essentiality also differs between growth conditions: 499 genes are essential on minimal media vs 346 on LB, reflecting the additional biosynthetic burden of minimal media.
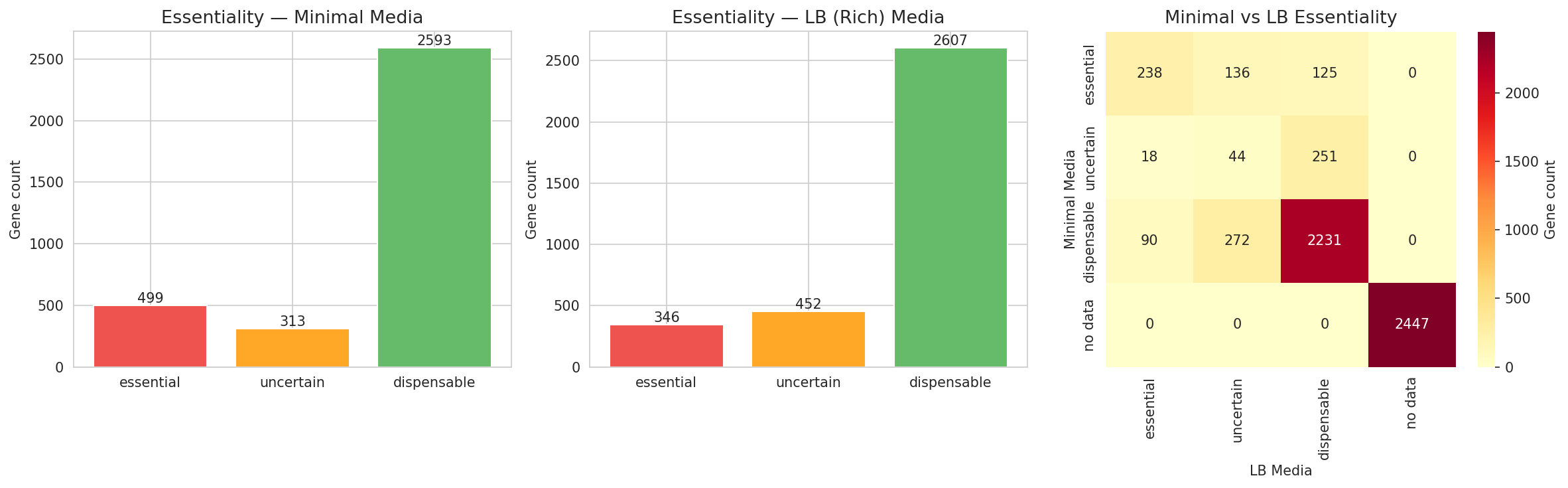
(Notebook: 04_gene_essentiality_and_fitness.ipynb)
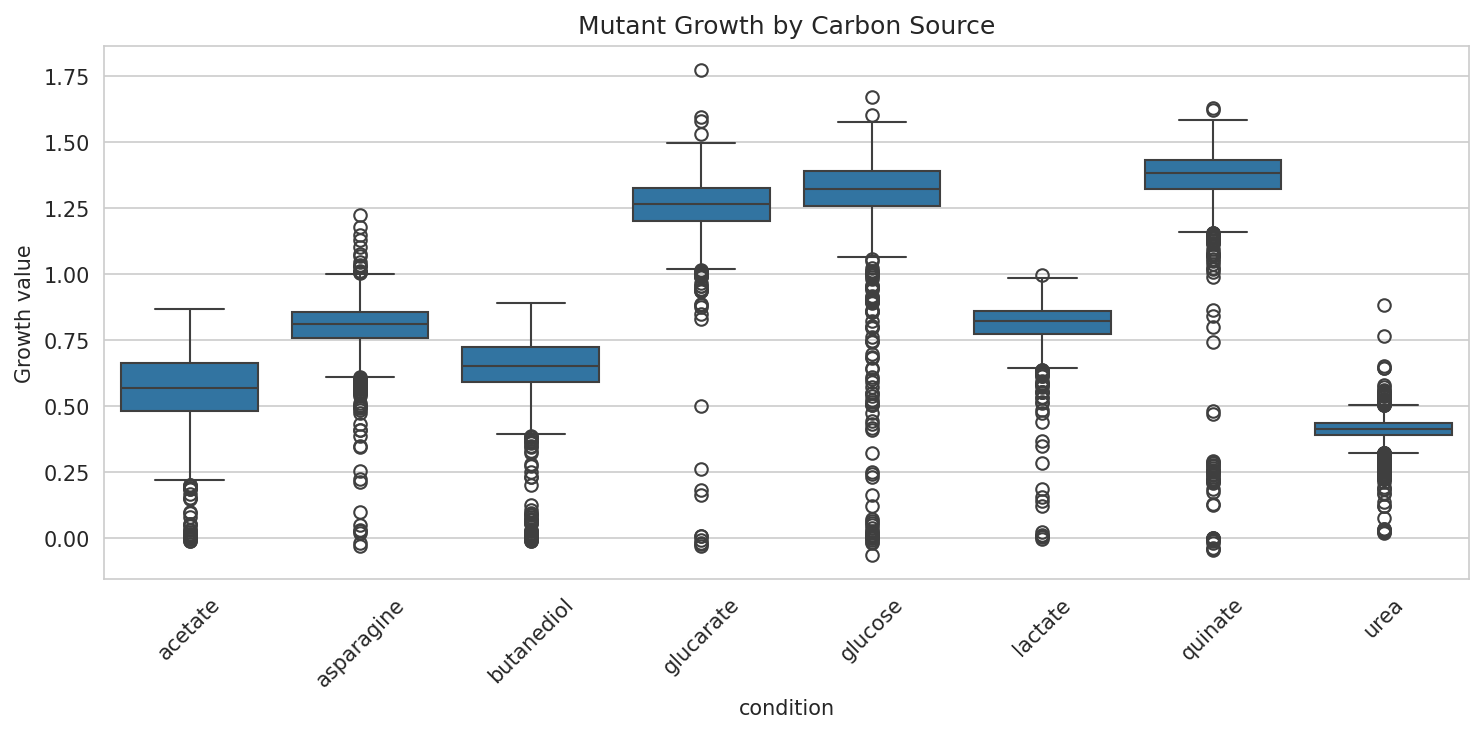
5. Condition-Specific Fitness: Urea and Quinate Stand Apart
Mutant growth fitness across 8 carbon sources shows moderate overall correlation (mean pairwise r = 0.44) but with notable outliers. Urea fitness is nearly uncorrelated with quinate (r = 0.11) and weakly correlated with all other conditions (r = 0.12-0.28), suggesting that urea catabolism involves a largely independent set of genes. Butanediol-acetate (r = 0.58) and butanediol-lactate (r = 0.53) show the strongest correlations, consistent with shared central carbon metabolism.
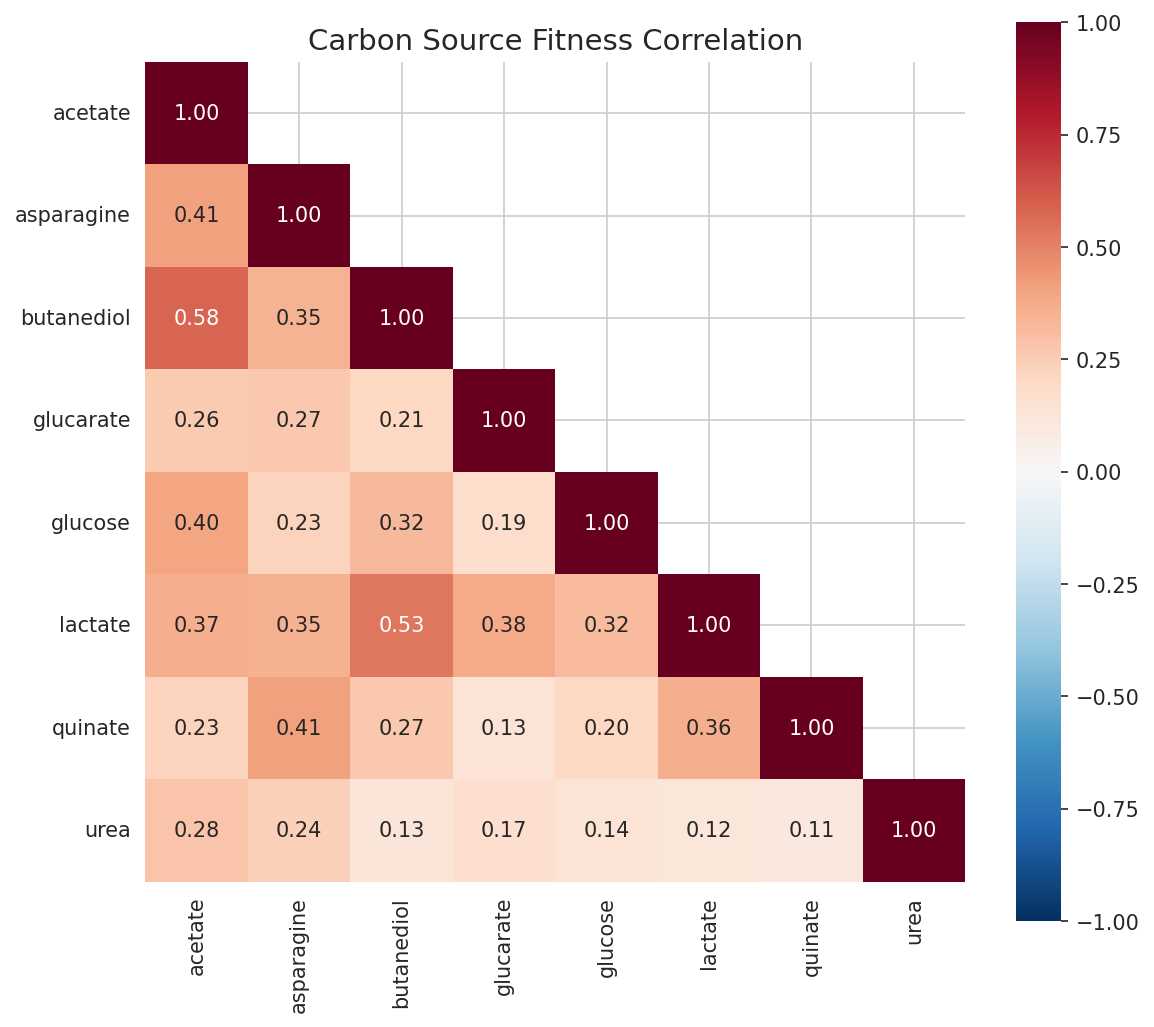
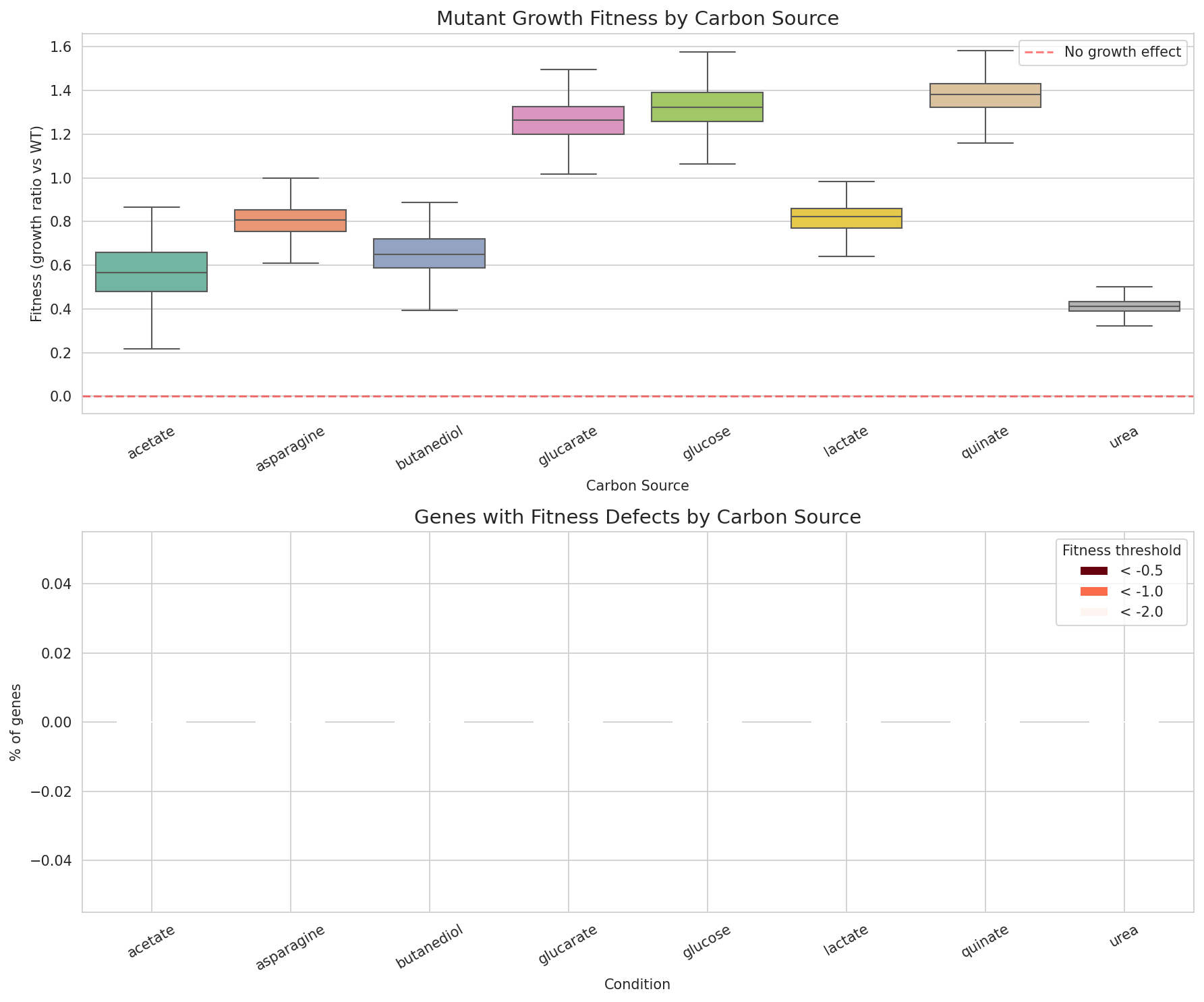
(Notebook: 04_gene_essentiality_and_fitness.ipynb)
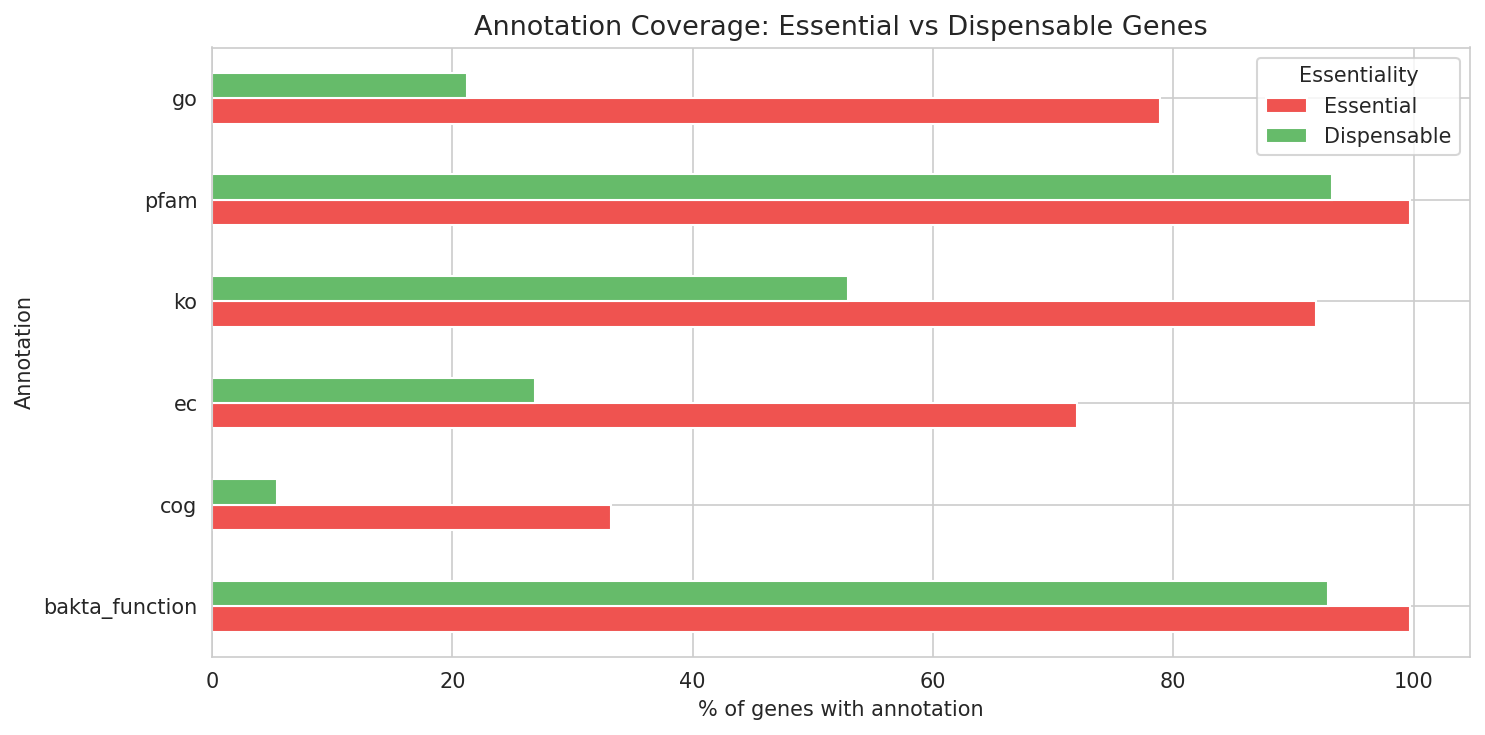
6. Essential Genes Are 6x More Likely to Have COG Annotations
Essential genes are dramatically more annotation-rich than dispensable genes: 33% of essential genes have COG assignments vs only 5% of dispensable genes. Similarly, 92% of essential genes have KEGG KO assignments vs 53% of dispensable genes. This pattern is expected — essential genes tend to encode well-characterized housekeeping functions — but it also means the functionally unannotated essential genes (~8% without KO) are particularly interesting as potential novel essential functions.
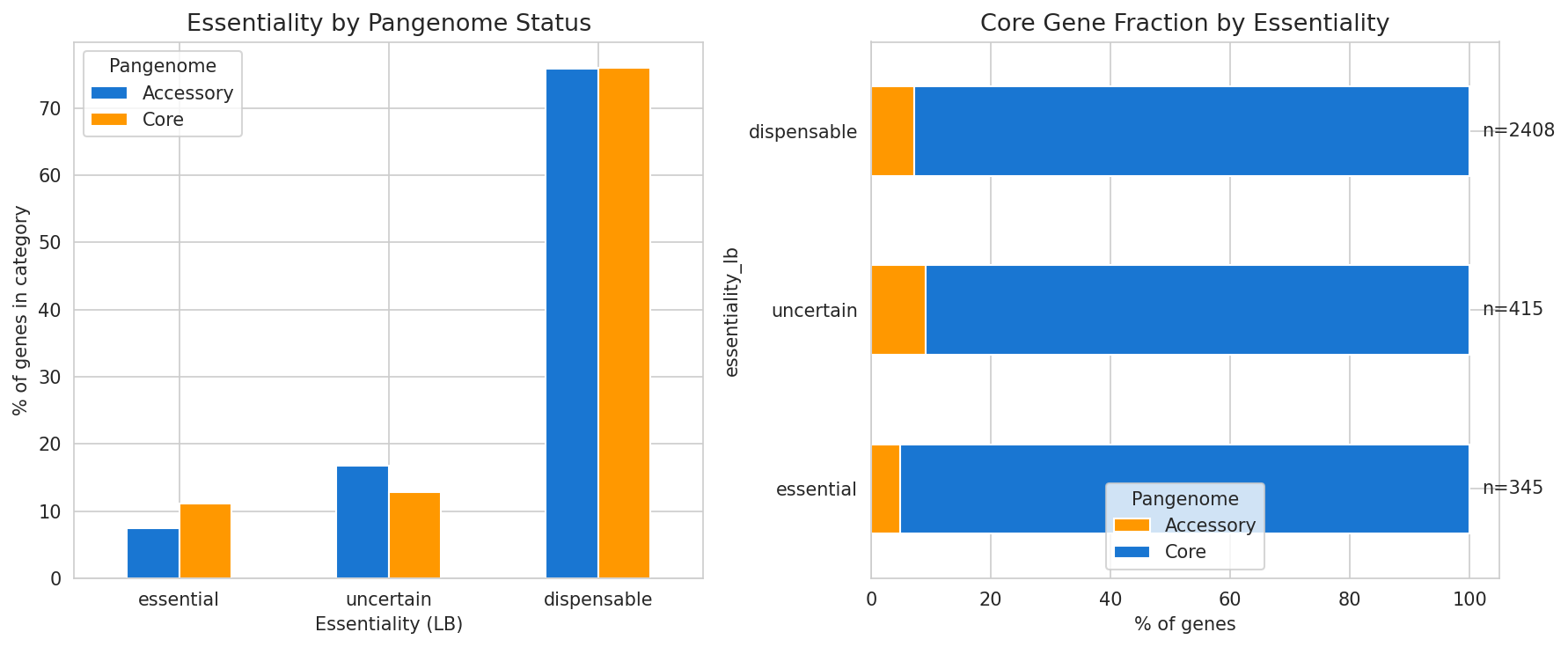
Essential genes are also more likely to be in the core pangenome, consistent with the pattern that conserved genes tend to be essential.
(Notebook: 04_gene_essentiality_and_fitness.ipynb)
7. Highly Conserved Core Metabolism Across 14 Genomes
Of 1,330 unique metabolic reactions, 1,248 (94%) are shared across all 14 Acinetobacter genomes (core), 62 are variable (present in 2-13 genomes), and only 20 are genome-unique. Gapfilling accounts for 7.7% of reactions on average, with 243 missing functions cataloged.
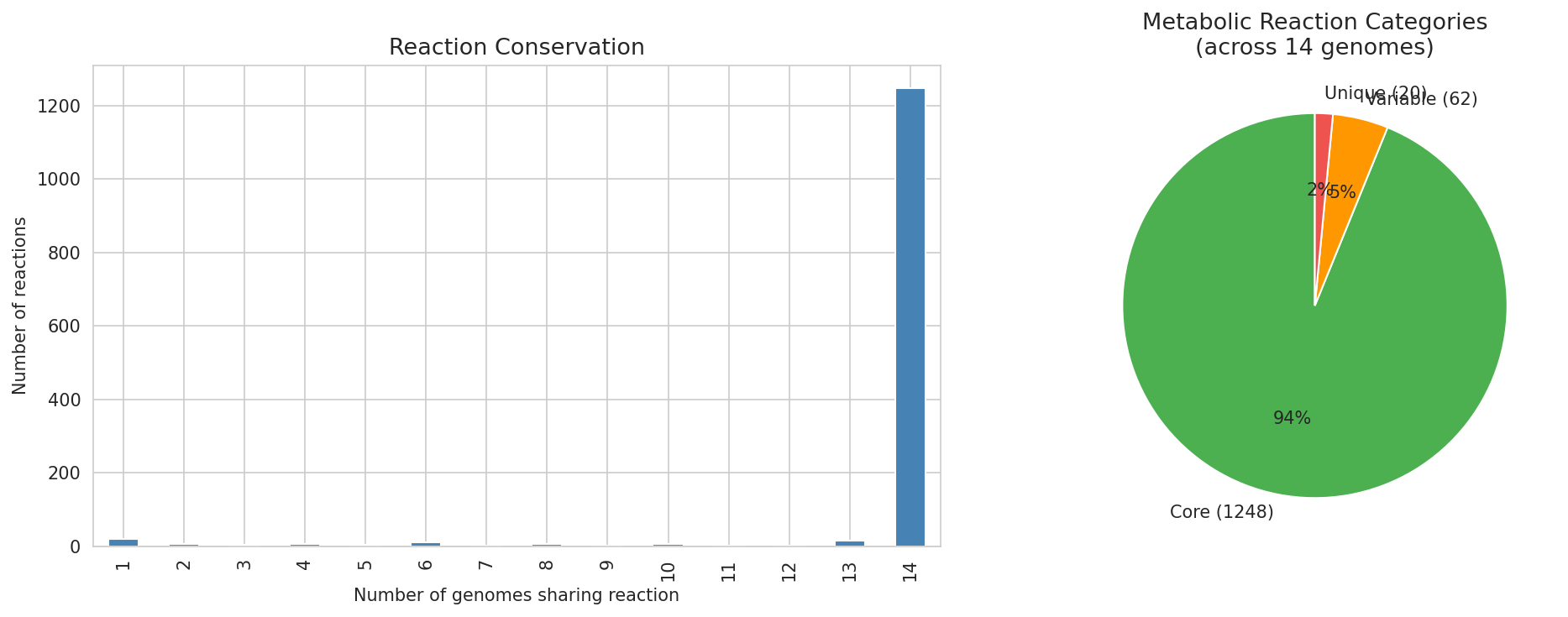
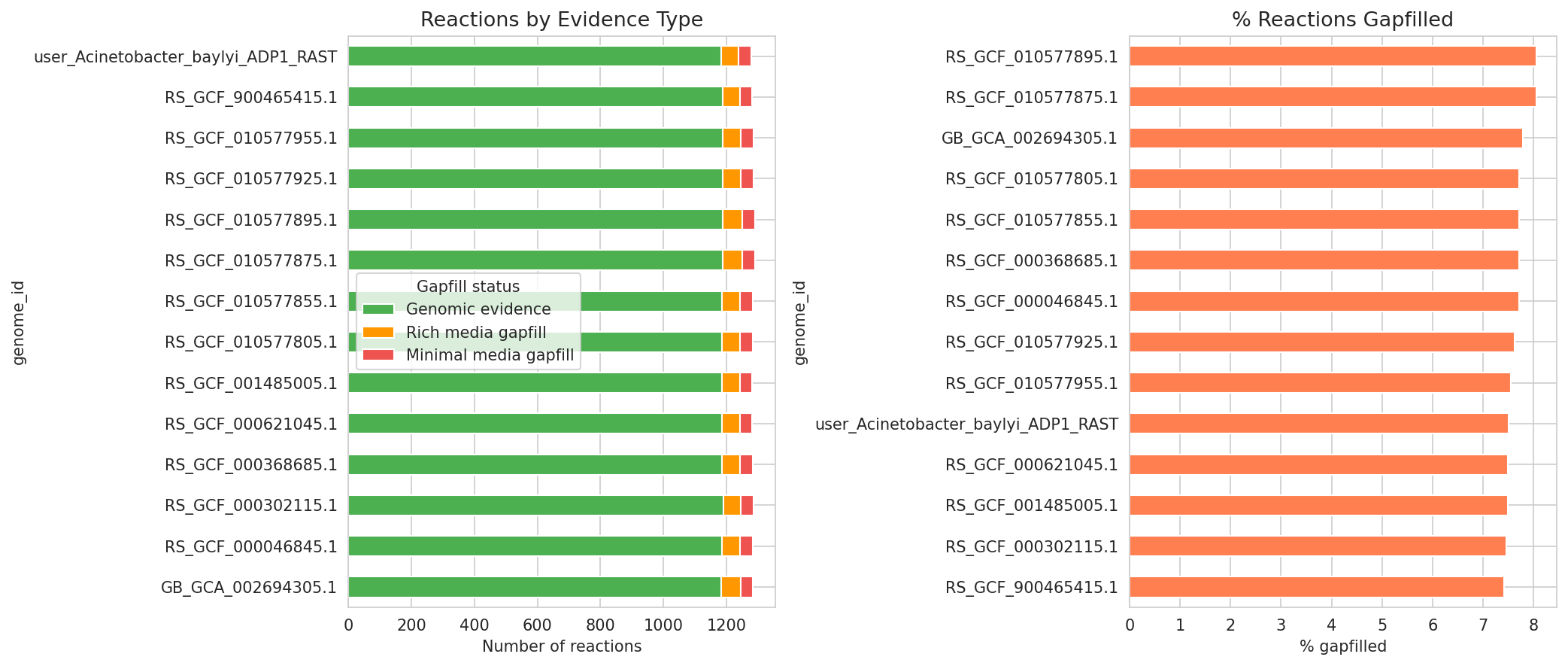
(Notebook: 05_metabolic_model_and_phenotypes.ipynb)
8. 87% of Growth Predictions Depend on Gapfilled Reactions
Of 121,519 growth phenotype predictions across 14 genomes, 105,376 (87%) require at least one gapfilled reaction. This heavy dependence on gapfilling means that prediction accuracy is tightly coupled to gapfilling quality. False negatives have higher mean gap counts than correct predictions.
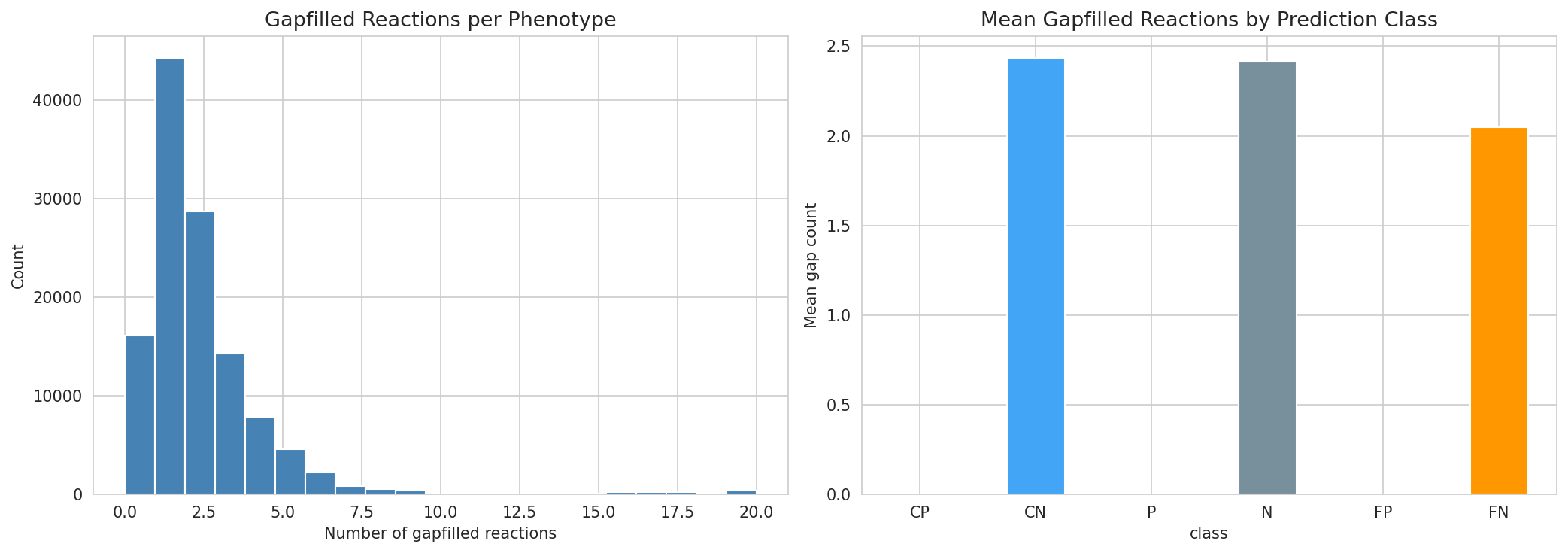
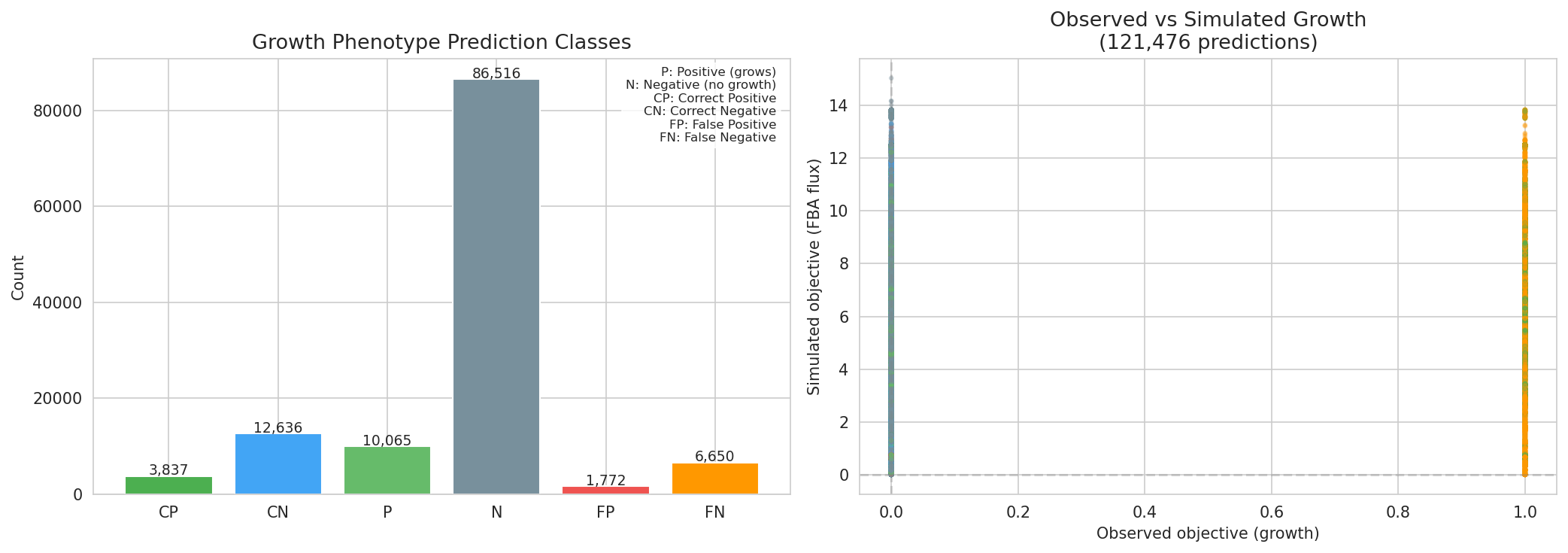
(Notebook: 05_metabolic_model_and_phenotypes.ipynb)
Results
Database Structure
The ADP1 database contains 15 interconnected tables with 27 relationship pairs (shared columns). The core data flow is: genome → genome_features (annotations, essentiality, proteomics, growth) → pan_genome_features (pangenome context) → genome_reactions (metabolic model) → gene_phenotypes (phenotype associations) → growth_phenotypes_detailed (FBA predictions).
Seven engineered ADP1 strains are documented in the strains table, with proteomics data available for all seven. The strains include wild-type ADP1 and six derivatives with aromatic amino acid pathway modifications (ΔaroF, ΔaroG, dgoA variants).
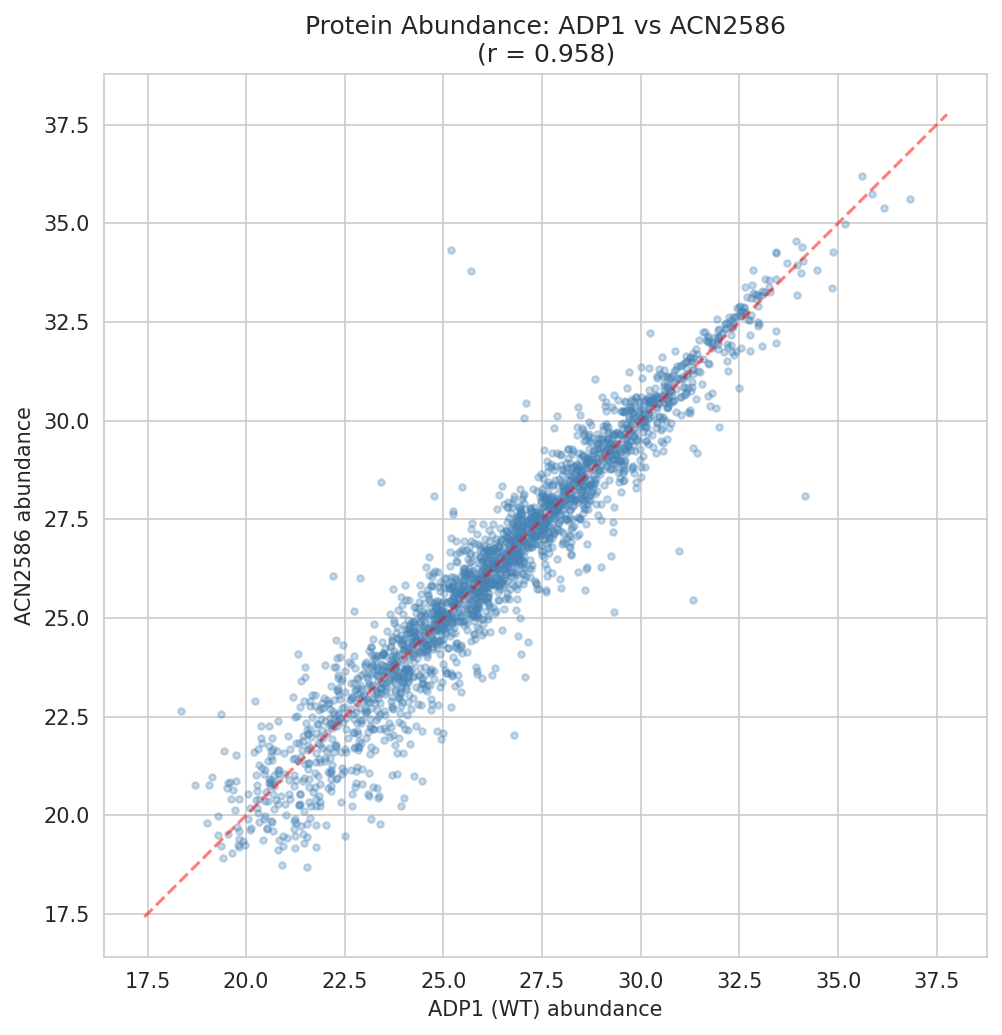
Proteomics Cross-Strain Analysis
Protein abundance was measured across 7 strains for 2,383 genes. Cross-strain correlation is high, indicating that the engineered modifications have targeted rather than global effects on the proteome.
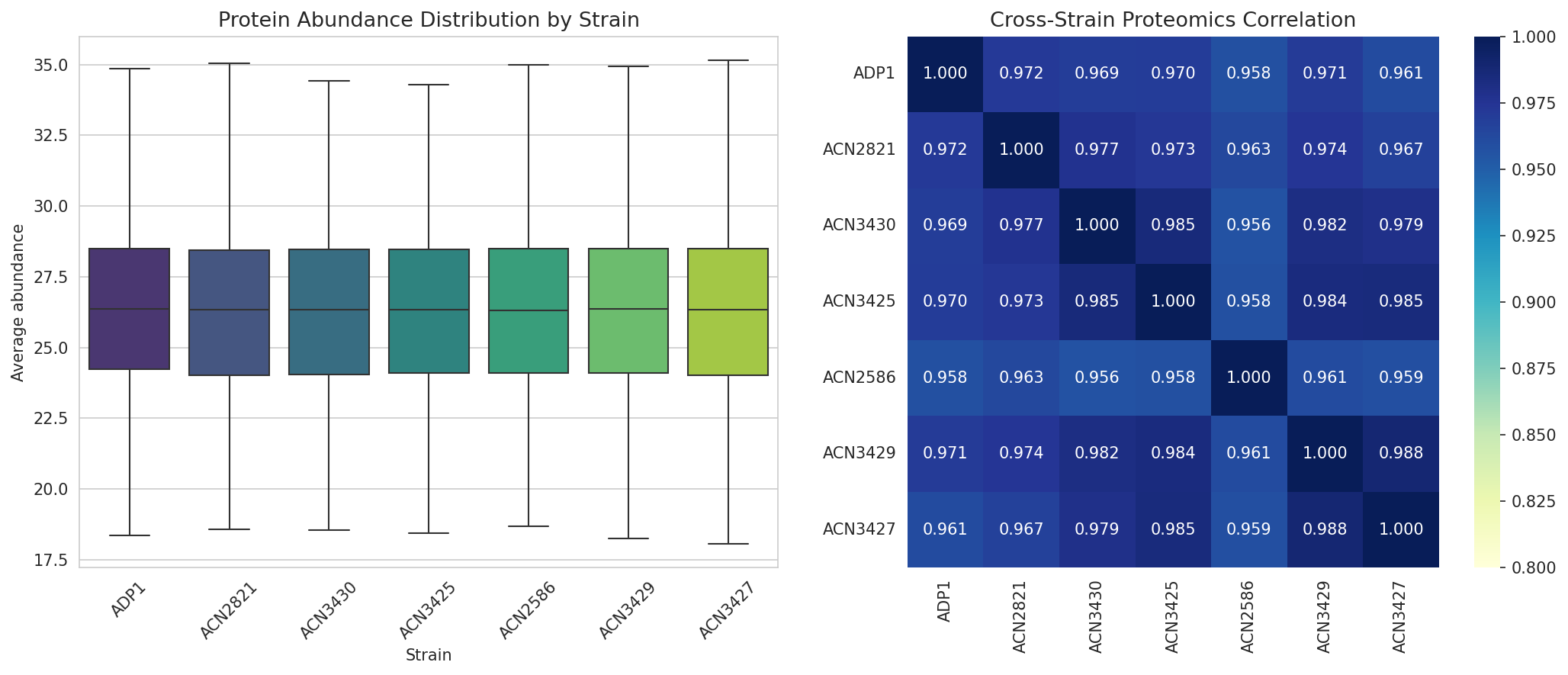
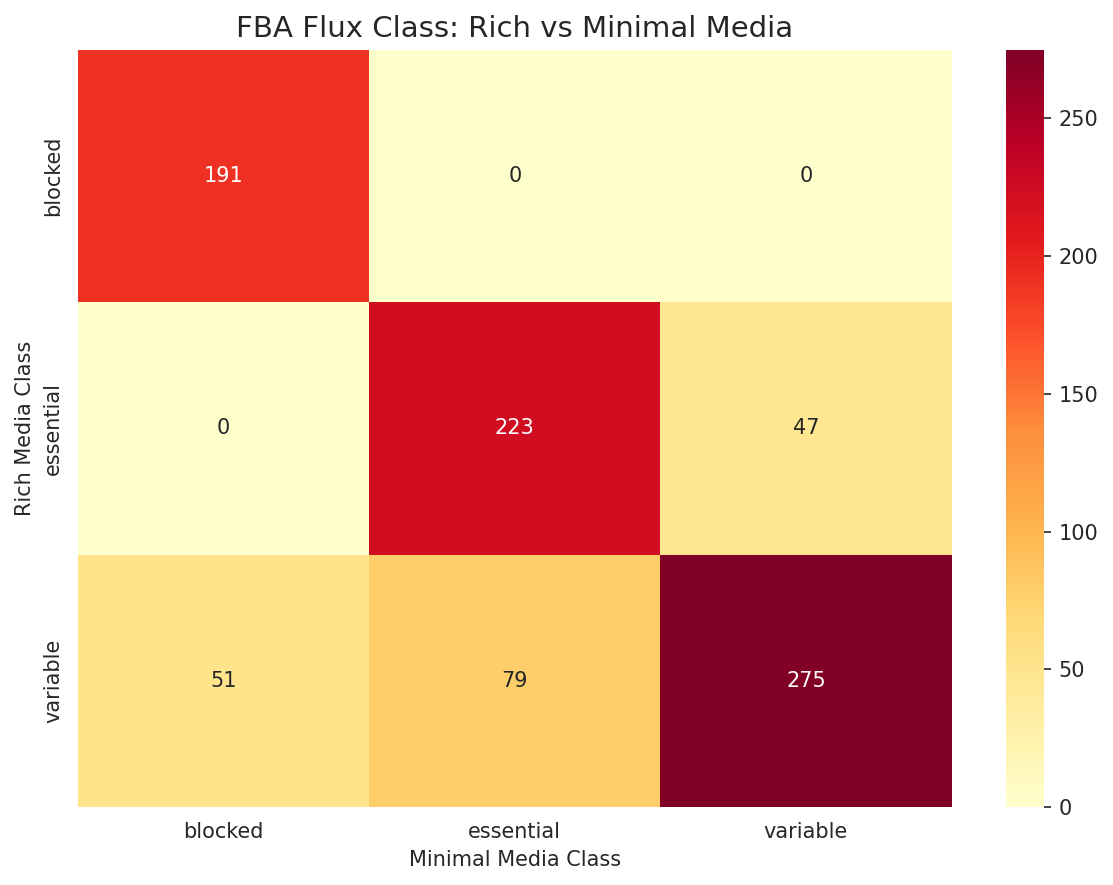
FBA Flux Class Transitions
20% of genes (177/866) change FBA flux class between rich and minimal media, reflecting condition-dependent metabolic rewiring.
Additional Figures
Interpretation
This exploration project demonstrates that a comprehensive user-provided database for A. baylyi ADP1 integrates deeply with BERDL collections. The four validated connection types (genome IDs, reactions, compounds, and pangenome clusters) together cover the major axes of biological data in the lakehouse — genomics, metabolomics, and comparative genomics.
Novel Contribution
The ADP1 database brings several data types not available elsewhere in BERDL:
- Mutant growth fitness on 8 carbon sources — ADP1 is absent from the Fitness Browser, making this the only source of condition-specific fitness data for this organism in the lakehouse
- Proteomics across 7 engineered strains — quantitative protein abundance data for wild-type and aromatic amino acid pathway mutants
- Gene essentiality calls — TnSeq-based essentiality on both minimal and LB media
- FBA model predictions — flux balance analysis with gapfilling metadata, enabling model-experiment concordance analysis
Limitations
- Data modality overlap: No gene has data across all 6 modalities. FBA flux data is the sparsest (15% of genes), limiting the concordance analysis to 866 genes.
- Pangenome cluster mapping is indirect: The BERDL ↔ ADP1 cluster mapping goes through 3 tables. While 100% complete, this indirection could introduce edge cases for clusters that split or merge between the two pangenome analyses.
- Gapfilling dependence: 87% of growth phenotype predictions rely on gapfilled reactions. The 243 missing functions represent genuine gaps in genomic evidence that affect prediction reliability.
- Single species: This database covers only A. baylyi. Cross-species comparisons would require similar databases for other organisms.
Future Directions
- Exploit the FBA-TnSeq discordance: The 227 genes where FBA and TnSeq disagree on essentiality could guide metabolic model refinement — are the discordant genes enriched for specific pathways or regulatory functions?
- Cross-species fitness comparison: Use the cluster ID mapping to compare ADP1 mutant growth data against Fitness Browser data for related organisms, even though ADP1 itself isn't in the FB.
- PhageFoundry integration: The 37-table Acinetobacter genome browser in PhageFoundry was identified but not deeply explored. Cross-referencing with phage susceptibility data could be valuable.
- Urea metabolism deep-dive: The near-zero correlation between urea fitness and other carbon sources (r = 0.11-0.28) suggests a largely independent gene set. Identifying these urea-specific genes and their pangenome conservation could be biologically interesting.
- Gapfilling quality assessment: With 243 missing functions and 87% of growth predictions depending on gapfilling, systematically assessing gapfill confidence using pangenome conservation data could improve prediction reliability.
Data
Sources
| Collection | Tables Used | Purpose |
|---|---|---|
User-provided berdl_tables.db |
All 15 tables | ADP1 genome, annotations, metabolic model, phenotypes, proteomics |
kbase_ke_pangenome |
genome, gene_cluster, gene_genecluster_junction, gtdb_species_clade, pangenome |
Pangenome context for A. baylyi (13 genomes, 4,891 clusters) |
kbase_msd_biochemistry |
reaction, molecule |
ModelSEED reaction/compound name resolution |
kescience_fitnessbrowser |
organism |
Checked for ADP1 presence (not found) |
phagefoundry_acinetobacter_genome_browser |
(37 tables) | Acinetobacter genome browser (identified, not deeply queried) |
Generated Data
| File | Rows | Description |
|---|---|---|
data/cluster_id_mapping.csv |
4,891 | BERDL ↔ ADP1 pangenome cluster ID mapping |
data/berdl_connection_summary.csv |
5 | Connection scan results summary |
References
- Parks DH, Chuvochina M, Rinke C, Mussig AJ, Chaumeil PA, Hugenholtz P. (2022). "GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy." Nucleic Acids Research. 50(D1):D199-D207.
- Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. (2010). "High-throughput generation, optimization and analysis of genome-scale metabolic models." Nature Biotechnology. 28(9):977-982.
- de Berardinis V, Vallenet D, Castelli V, Besnard M, Pinet A, Cruaud C, Samair S, Lechaplais C, Gyapay G, Riber C, et al. (2008). "A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1." Molecular Systems Biology. 4:174.
- Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, et al. (2018). "KBase: The United States Department of Energy Systems Biology Knowledgebase." Nature Biotechnology. 36(7):566-569.
Data Collections
Review
Summary
This is a well-executed exploratory project that systematically characterizes a 136 MB user-provided SQLite database for Acinetobacter baylyi ADP1 and validates its integration points with four BERDL collections. The project excels in documentation quality, visual richness (18 figures across 5 notebooks), and methodological transparency — the cluster ID mapping problem (0% direct string match → 100% gene-level bridge) is particularly well handled. The three-file structure (README, RESEARCH_PLAN, REPORT) follows observatory conventions, all notebooks have saved outputs, and the REPORT presents 8 clearly numbered findings with supporting figures. The main areas for improvement are minor: the reproduction instructions cover only one notebook, the PhageFoundry scan cell produced no output, and there are a few opportunities for deeper statistical analysis.
Methodology
Research question: Clearly stated as a two-part exploration: (1) characterize the database, (2) identify BERDL connection points. The framing as an exploration project rather than a hypothesis-driven study is appropriate and honestly acknowledged in the RESEARCH_PLAN.
Approach: The phased approach (inventory → connection scan → integration deep-dives) is logical and well-matched to the question. The research plan's connection mapping table (RESEARCH_PLAN.md lines 20-29) lays out 8 identifier types with specific BERDL targets and link strategies, providing a clear checklist.
Data sources: Well documented in both README and REPORT. The README identifies 6 data sources (user-provided + 5 BERDL collections), and the REPORT's Data section includes a table showing which tables were used from each collection and their purpose. The identification of ADP1's absence from the Fitness Browser (NB02 cell 12) is a useful negative result.
Reproducibility:
- Notebook outputs: Excellent — 58 of 59 code cells across all 5 notebooks have saved outputs. The one exception is cell 15 in NB02 (PhageFoundry ADP1/baylyi search), which produced no output — likely because the search found no matches, but this should be documented with a print statement.
- Figures: Outstanding — 18 figures in the figures/ directory, covering every major analysis stage (database inventory, BERDL connections, cluster mapping, essentiality, fitness, proteomics, metabolic model, gapfilling, growth predictions, annotations, ontology). Each figure is referenced in the REPORT with a description.
- Dependencies: requirements.txt is present with 3 packages (pandas, matplotlib, seaborn). However, it omits the berdl_notebook_utils dependency used in NB02 and NB03 for Spark access.
- Reproduction guide: The README has a ## Reproduction section with prerequisites and a run command, but it only shows how to execute NB01 (01_database_exploration.ipynb). The guide should cover all 5 notebooks and note that NB02 and NB03 require Spark access via the BERDL JupyterHub environment.
- Spark/local separation: Partially documented. NB02 and NB03 use Spark; NB01, NB04, and NB05 use only SQLite and can run locally. This distinction is not explicitly called out in the README. The cached outputs (data/cluster_id_mapping.csv, data/berdl_connection_summary.csv) enable downstream work without Spark re-execution.
Code Quality
SQL correctness: SQLite queries throughout NB01 are straightforward and correct. The BERDL Spark queries in NB02 use proper IN clause batching (batch_size=100 for reactions, batch_size=200 for cluster junctions), avoiding performance issues with overly large IN lists. The seed.reaction: prefix handling for biochemistry lookups (NB02 cell 6) is correct.
Statistical methods: Appropriate for an exploratory project. Pearson correlation for cross-strain proteomics and carbon source fitness, cross-tabulation for essentiality concordance, and simple descriptive statistics throughout. The concordance analysis (NB04 cell 5) correctly computes agreement rate. The FBA essentiality definition (rich_media_class == 'essential') is reasonable but could be documented more explicitly — the mapping between FBA flux classes and essentiality is a modeling choice.
Notebook organization: Each notebook follows a clean structure: markdown header with goals → imports/setup → numbered analysis sections → summary. NB04 is the most complex (8 sections, 10 code cells) and handles it well with clear section headers.
Pitfall awareness:
- The project correctly handles the pangenome cluster ID naming convention difference (a known pitfall pattern from docs/pitfalls.md regarding different ID formats between systems).
- The gene_genecluster_junction queries use appropriate batching (pitfall: this is a large table).
- The project does not encounter the -- in species IDs pitfall because it uses exact equality with proper quoting in NB02 cell 4.
- No issues with string-typed numeric columns (the SQLite database appears to use proper types).
Minor code issues:
1. In NB04 cell 5, the FBA concordance defines "essential" as rich_media_class == 'essential' but the actual flux class values include essential_forward and essential_reverse (visible in NB01 cell 19). The concordance calculation may undercount FBA-essential genes if the check doesn't match these class names — worth verifying.
2. In NB04 cell 9, the correlation mask is np.triu(..., k=1) (upper triangle), but the upper = corr.where(~mask).stack() extracts the lower triangle. This is mathematically correct (lower triangle = upper triangle values by symmetry) but the variable name upper is misleading.
Findings Assessment
Conclusions supported by data: All 8 key findings in the REPORT are directly supported by notebook outputs. The connection scan results (100% genome, 91% reaction, 100% compound, 100% cluster mapping) are verifiable in NB02 and NB03. The FBA-TnSeq concordance of 73.8% is computed and shown in NB04. The gapfilling dependency (87%) is computed in NB05.
Limitations acknowledged: Yes — the REPORT's Limitations section (lines 139-143) identifies four specific limitations: sparse data overlap, indirect cluster mapping, gapfilling dependence, and single-species scope. These are honest and relevant.
Incomplete analysis:
- The PhageFoundry connection (NB02 cells 14-15) was identified but not explored. Cell 14 lists 37 tables and shows 15 table names, but cell 15 (ADP1/baylyi search) produced no output. The REPORT acknowledges this as a future direction.
- The UniRef connections (3,100+ IDs identified in NB01) were not validated against BERDL in NB02, despite being listed as a connection point. This is a gap — the project reports 4 of 5 connection types validated, but UniRef, COG, KEGG KO, EC, and Pfam connections were identified but not tested.
- The growth_phenotype_summary table shows all genomes with 0.0 accuracy and zero true/false positives/negatives (NB01 cell 24), which suggests the observed growth data may be missing for most genomes. This is not discussed.
Visualizations: Clear and properly labeled throughout. Figures use consistent color palettes (red for essential, green for dispensable, blue for core), include titles, axis labels, and annotations (counts on bars, correlation values). The proteomics scatter plot (NB04 cell 12) and growth correlation heatmap (NB04 cell 9) are particularly effective.
Suggestions
-
Expand reproduction instructions (medium priority): Update the README's Reproduction section to list all 5 notebooks with their execution order, note which require Spark (NB02, NB03) vs local-only (NB01, NB04, NB05), and include expected runtimes. Currently only NB01 is shown.
-
Add a print statement to the empty PhageFoundry cell (low priority): NB02 cell 15 produces no output. Add a summary line like
print(f'Searched {len(pf_tables)} tables, found N matches')so readers can confirm the search completed. -
Validate UniRef connections (medium priority): The project identifies 3,100+ UniRef IDs as connection points (NB01 cell 31) but never tests them against BERDL in NB02. Even a sample check of 100 UniRef50 IDs against
kbase_uniref50would strengthen the connection inventory. -
Investigate the zero-accuracy growth phenotype summary (medium priority): NB01 cell 24 shows all genomes with 0.0 accuracy and zero confusion matrix values in
growth_phenotype_summary. This likely means observed growth data was not available for accuracy calculation, but it's worth noting in the REPORT since the growth phenotype analysis in NB05 presents prediction classes without discussing validation. -
Clarify FBA essentiality definition (low priority): In NB04 cell 5, verify that
rich_media_class == 'essential'is the correct filter — the flux class values shown in NB01 cell 19 areessential_forwardandessential_reverse, not bareessential. If the filter is checking for a different column value than what appears in the data, the concordance numbers may need correction. -
Add
berdl_notebook_utilsto requirements.txt or note the Spark dependency (low priority): NB02 and NB03 import fromberdl_notebook_utils.setup_spark_session, which is only available on the BERDL JupyterHub. Therequirements.txtshould either include this or note that NB02/NB03 require the BERDL environment. -
Consider adding a combined data coverage UpSet plot (nice-to-have): NB04 cell 18 shows that no gene has data across all 6 modalities. An UpSet plot showing the intersection sizes across modalities would more clearly convey the pairwise and higher-order overlaps mentioned in Finding 1.
This review was generated by an AI system. It should be treated as advisory input, not a definitive assessment.
Visualizations
Annotation By Essentiality
Data Coverage By Modality
Essentiality Distribution
Essentiality Overview
Essentiality Vs Pangenome
Fba Flux Class Transition
Fba Tnseq Concordance
Gapfilling Impact
Growth Condition Correlation
Growth Phenotype Predictions
Metabolic Flux Classes
Mutant Growth By Condition
Mutant Growth Fitness
Ontology Distribution
Proteomics Cross Strain
Proteomics Wt Vs Engineered
Reaction Conservation
Reaction Gapfilling
Notebooks
01_database_exploration.ipynb
01 Database Exploration
View notebook →
02_berdl_connection_scan.ipynb
02 Berdl Connection Scan
View notebook →
03_cluster_id_mapping.ipynb
03 Cluster Id Mapping
View notebook →
04_gene_essentiality_and_fitness.ipynb
04 Gene Essentiality And Fitness
View notebook →
05_metabolic_model_and_phenotypes.ipynb
05 Metabolic Model And Phenotypes
View notebook →
Data Files
| Filename | Size |
|---|---|
berdl_connection_summary.csv |
0.3 KB |
cluster_id_mapping.csv |
309.4 KB |